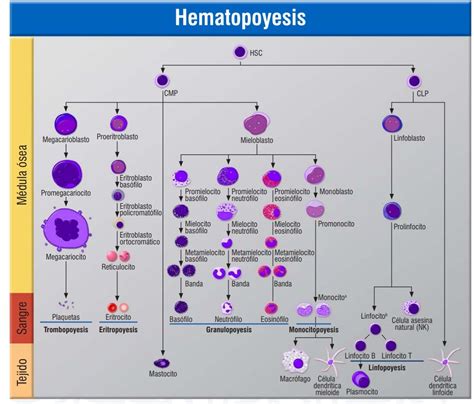
La hematopoyesis (o hemopoyesis) es el proceso fundamental de producción de las células sanguíneas, que abarca la proliferación, diferenciación y maduración celular. Este proceso se localiza en diferentes sitios a lo largo de la vida, iniciando en el saco vitelino durante las fases tempranas del embrión (fase mesoblástica) y luego migrando al hígado (fase hepática) alrededor de la segunda semana de gestación. Aproximadamente en la 11.ª semana, las células hematopoyéticas colonizan la médula ósea, que se convierte en el principal punto de formación celular. En los adultos, después de los 20 años, la hematopoyesis de las tres principales líneas celulares se concentra en el tejido medular de los huesos planos.
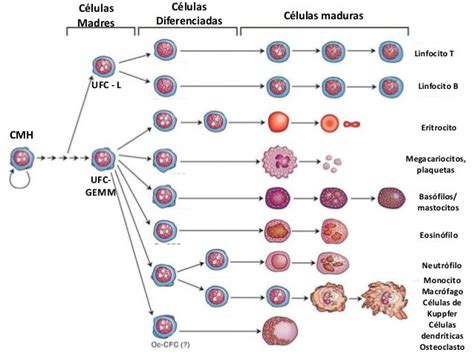
La Célula Madre Hematopoyética y su Diferenciación
Todos los tipos de células sanguíneas (eritrocitos, leucocitos y plaquetas) se originan de un precursor común: la célula madre hematopoyética (citoblasto pluripotencial). Estas células poseen la capacidad única de colonizar la médula ósea y reconstituir el sistema hematopoyético en caso de destrucción. Al diferenciarse, las células madre hematopoyéticas dan origen a dos líneas celulares principales:
- Células mieloides: precursores de eritrocitos, granulocitos, monocitos y plaquetas.
- Células linfoides: precursores de linfocitos.
Las células mieloides y linfoides, conocidas como células progenitoras multipotenciales, tienen un potencial de diferenciación menor que las células madre hematopoyéticas. Al dividirse, pueden formar nuevas células progenitoras para mantener su población o diferenciarse en células precursoras, denominadas blastos, donde se observan las características distintivas de cada línea celular.
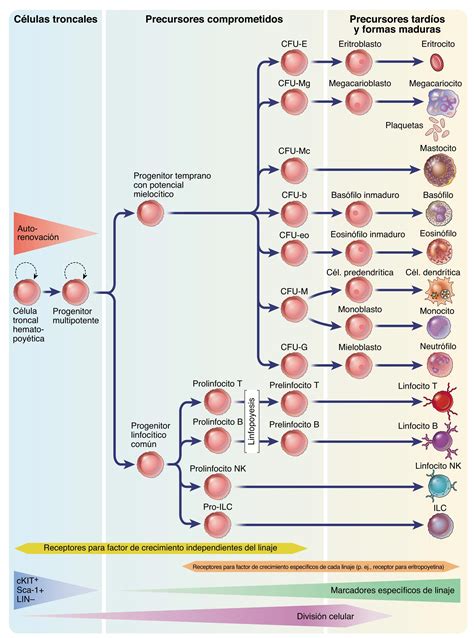
Formación de Eritrocitos (Eritropoyesis)
La eritropoyesis, el proceso de formación de los eritrocitos, inicia a partir de una célula madre hematopoyética. El principal estímulo para la formación de eritrocitos es la eritropoyetina, una hormona secretada por los riñones en respuesta a la reducción de oxígeno en la sangre.
Etapas de Maduración de los Eritrocitos:
- Proeritroblastos: Células grandes con citoplasma basófilo y núcleo grande con cromatina no condensada.
- Eritroblastos basófilos: Bajo la influencia de la eritropoyetina, el citoplasma se vuelve intensamente basófilo.
- Eritroblastos ortocromatófilos (normoblastos): El volumen celular disminuye, se pierde la basofilia y las células se vuelven completamente acidófilas.
- Reticulocitos: Los normoblastos expulsan su núcleo, dando origen a estas células anucleadas.
Formación de Granulocitos (Granulocitopoyesis)
La granulocitopoyesis es el proceso de maduración de los granulocitos, que se caracterizan por la presencia de gránulos azurófilos y específicos. Incluyen neutrófilos, basófilos y eosinófilos. Los neutrófilos son cruciales en el combate de infecciones bacterianas, mientras que los gránulos de los basófilos contienen histamina y otros agentes inflamatorios.
Etapas de Maduración de los Granulocitos:
- Mieloblasto: Célula inmadura con gran núcleo, nucléolos y citoplasma basófilo con gránulos azurófilos.
- Promielocito: Célula más pequeña y basófila con núcleo esférico o dentado.
- Mielocito: El citoplasma pierde basofilia y adquiere gran cantidad de gránulos específicos, diferenciándose en mielocitos neutrófilos, basófilos o eosinófilos.
- Metamielocito: Núcleo con hendidura profunda, indicando el inicio de la lobulación.
- Granulocitos con núcleos en bastón (bastonetes): Núcleos alargados. En esta fase, solo los neutrófilos son comúnmente caracterizados.
- Granulocitos maduros: El núcleo de los neutrófilos se segmenta en 2 o 3 lóbulos.
Formación de Plaquetas (Trombopoyesis)
La trombopoyesis (también conocida como trombocitopoyesis o megacariocitopoyesis) es el proceso de formación de las plaquetas.
Etapas de Maduración de las Plaquetas:
- Megacarioblasto: Célula inmadura con núcleo grande, numerosos nucléolos y citoplasma basófilo.
- Megacariocito: Células grandes con núcleo lobulado y citoplasma menos basófilo, conteniendo gránulos que darán origen a los cromómeros plaquetarios.
Formación de Linfocitos
Los linfocitos se originan a partir de células de la línea linfoide.
Etapas de Maduración de los Linfocitos:
- Linfoblasto: Célula relativamente grande, redondeada, con citoplasma basófilo.
- Prolinfocito: Célula más pequeña con citoplasma basófilo y posibles gránulos azurófilos.
La maduración de los linfocitos T ocurre en el timo, y la de los linfocitos B, en la médula ósea.
Alteraciones Hematológicas en el Adulto Mayor
Las alteraciones hematológicas pueden manifestarse como una reducción, un aumento o una disfunción de las células sanguíneas maduras o de una línea celular completa. El sufijo "penia" denota una reducción en el número total de células (ej., trombocitopenia para plaquetas, linfopenia para linfocitos).
Síndromes Mielodisplásicos (SMD)
Los síndromes mielodisplásicos son un grupo de trastornos clonales de células madre hematopoyéticas que se caracterizan por citopenia periférica, progenitores hematopoyéticos displásicos, médula ósea hipercelular y un alto riesgo de progresión a leucemia mieloide aguda (LMA). Estos síndromes se diagnostican con mayor frecuencia en personas en la octava década de vida, con un riesgo que aumenta con la edad debido a la acumulación de mutaciones somáticas y la exposición a toxinas ambientales.
Comprender los síndromes mielodisplásicos (SMD)
Fisiopatología del Síndrome Mielodisplásico
Los SMD son trastornos de células madre hematopoyéticas clonales unidos por mutaciones distintivas, frecuentemente en genes implicados en el empalme del ARN. Se caracterizan por una hematopoyesis ineficaz y displásica, que puede resultar en anemia (la más común), neutropenia o trombocitopenia. En algunos casos, se puede observar hematopoyesis extramedular, que lleva a hepatomegalia y esplenomegalia.
Clasificación de los Síndromes Mielodisplásicos:
La clasificación de los SMD depende de los hallazgos en sangre, médula ósea, cariotipo y mutaciones:
- Anemia refractaria: Anemia con reticulocitopenia; médula normal o hipercelular con hiperplasia eritroide y diseritropoyesis; blastos ≤ 5% de las células nucleadas de la médula ósea.
- Anemia refractaria con sideroblastos en anillo: Similar a la anemia refractaria, pero con > 15% de sideroblastos en anillo.
- Citopenia refractaria con displasia multilinaje: Citopenia no restringida a los eritrocitos; displasia prominente de precursores de leucocitos y megacariocitos.
- Citopenia refractaria con displasia multilinaje y sideroblastos en anillo: Igual que la citopenia refractaria con displasia multilinaje, pero con > 15% de sideroblastos en anillo.
- Anemia refractaria con exceso de blastos: Citopenia de ≥ 2 líneas celulares con anomalías morfológicas; médula hipercelular con diseritropoyesis y disgranulopoyesis; blastos del 5 al 19% de las células nucleadas de la médula ósea.
- Síndrome mielodisplásico, no clasificado: SMD que no encaja en ninguna categoría definida.
- Síndrome mielodisplásico con el defecto aislado del(5q): Anemia y trombocitosis típicamente graves, con deleción del brazo largo del cromosoma 5.
- Leucemia mielomonocítica crónica (LMMC) y leucemia mielomonocítica juvenil (LMMJ): Enfermedades mielodisplásicas/neoplasias mieloproliferativas mixtas con monocitosis absoluta en sangre y aumento de precursores de monocitos en médula ósea.
- Leucemia neutrofílica crónica: Caracterizada por neutrofilia y ausencia del cromosoma Filadelfia y del gen de fusión BCR-ABL1.
Signos y Síntomas de los Síndromes Mielodisplásicos
Los síntomas reflejan la línea celular más afectada:
- Anemia: Palidez, debilidad, cansancio.
- Neutropenia: Fiebre, infecciones recurrentes.
- Trombocitopenia: Aumento de hematomas, petequias, epistaxis, hemorragia mucosa.
También son comunes la esplenomegalia y la hepatomegalia. La médula ósea puede ser hipocelular o hipercelular. La evolución del clon del SMD tiende a la leucemia mieloide aguda.
Diagnóstico de los Síndromes Mielodisplásicos
Se sospecha SMD en pacientes (especialmente adultos mayores) con anemia, leucopenia o trombocitopenia resistentes al tratamiento.
El diagnóstico se realiza mediante:
- Hemograma completo.
- Frotis de sangre periférica: Revela macrocitosis, anisocitosis en anemia; plaquetas de tamaño variable, algunas hipogranulosas en trombocitopenia; neutrófilos con granularidad disminuida, anisocitosis, pseudo-células de Pelger-Huët; monocitosis en LMMC.
- Examen de médula ósea: Aspiración y biopsia para evaluar anormalidades morfológicas y citogenéticas.
Es fundamental descartar otras causas de citopenias, como trastornos autoinmunitarios, deficiencias vitamínicas, anemia aplásica idiopática, etc. El síndrome de deleción 5q-, una forma única de SMD, se caracteriza por anemia macrocítica y trombocitosis, respondiendo bien a la lenalidomida.
Tratamiento de los Síndromes Mielodisplásicos
El tratamiento se reserva generalmente para pacientes sintomáticos e incluye:
- Mejoría sintomática y tratamiento de soporte: Transfusiones crónicas de sangre y plaquetas. La quelación de hierro puede ser útil en pacientes con sobrecarga de hierro.
- Estimulantes de eritrocitos (EE): Reducen la gravedad de la anemia en un 15-20% de los pacientes, especialmente en aquellos no dependientes de transfusiones con eritropoyetina sérica < 500 mUI/mL. El luspatercept ha mostrado ser eficaz en pacientes con SMD de riesgo muy bajo a intermedio con sideroblastos en anillo.
- Fármacos moduladores epigenéticos:
- Azacitidina: Análogo de nucleósido pirimidina que prolonga la supervivencia general.
- Decitabina: También análogo de nucleósido pirimidina, induce remisión en un 43% de los pacientes.
- Lenalidomida: Inmunomodulador eficaz para reducir la dependencia de transfusiones en pacientes con síndrome de deleción 5q-.
- Inmunosupresión: Con ciclosporina con o sin globulina antitimocítica (ATG) para SMD hipoplásico.
- Trasplante de células madre hematopoyéticas alogénicas: Es el único tratamiento curativo y se indica en pacientes más jóvenes en buen estado físico, generalmente en grupos de riesgo intermedio 2 y alto riesgo.
Pronóstico de los Síndromes Mielodisplásicos
El pronóstico depende de la clasificación y trastornos asociados. Los pacientes con síndrome de deleción 5q-, anemia resistente al tratamiento o anemia resistente al tratamiento con sideroblastos en forma de anillo tienen una menor probabilidad de progresar a formas más agresivas.
El Revised International Prognostic Scoring System (IPSS-R) predice la evolución considerando:
- Citogenética: Peor pronóstico con anomalías múltiples o de alto riesgo.
- Porcentaje de blastos de la médula ósea: Peor pronóstico con mayor número de blastos (particularmente > 10%).
- Grado de citopenia: Peor pronóstico asociado con hemoglobina < 8 g/dL, recuento de plaquetas < 50.000/mcL y recuento absoluto de neutrófilos < 800/mcL.
Un mayor número de factores de riesgo indica un peor pronóstico, con medianas de supervivencia global que varían desde 0,8 años para el grupo de mayor riesgo hasta 8 años para el de menor riesgo.
Hematopoyesis Extramedular (HEM)
La hematopoyesis extramedular (HEM) es la presencia de células hematopoyéticas progenitoras con localización fuera de la médula ósea. Es característica de la vida intrauterina, pero en adultos se considera un mecanismo compensador en pacientes con anemia crónica o puede presentarse en el curso de hemoglobinopatías, anemias hemolíticas, leucemias, enfermedades mieloproliferativas, linfomas o asociada a pancitopenia iatrogénica.
Localizaciones y Presentaciones Clínicas de la HEM
Hígado, bazo y ganglios linfáticos son frecuentemente implicados. Sin embargo, la HEM puede desarrollarse en otras localizaciones, a menudo asintomáticamente. Cuando la HEM es sintomática (HEMS), no debe ser considerada un mecanismo compensador, sino una verdadera entidad clínica con una gran variedad de cuadros clínicos, según el órgano afectado:
- HEMS en SNC y periférico: Cefalea intratable, afectación cerebelar, afasia, hemiparesia, pérdida del control esfintérico, compresión medular, ciatalgia, exoftalmos monolateral, hemorragia subdural o paraparesia-paraplejía.
- HEMS torácica: Fibrosis pulmonar, derrame pleural, hemotórax, masa mediastinal o intratorácica, e infiltración cardíaca (precordialgia, disnea cardíaca, taponamiento o masa intraauricular).
- HEMS abdominal: Infiltración peritoneal (ascitis severa o tejido proliferante), cuadros de uropatía obstructiva, infiltración tubulointersticial, urolitiasis, síndrome nefrótico, glomerulonefritis, insuficiencia renal aguda o masas de dudoso significado.
- HEMS intestinal: Hemorragias, estreñimiento importante, oclusión o abdomen agudo quirúrgico.
- HEMS cutánea: Nódulos, lesiones papulares rojo-violetas, eritema papular o difuso, lesiones ulceradas, placas eritematosas, ampollas.
- Otras localizaciones: Tiroides, suprarrenal, testículo, próstata, ovario, endometrio y oído medio.
En un caso reportado, la infiltración meningoencefálica de la HEM en un paciente con linfoma no Hodgkin, en lugar de ser un mecanismo compensador, desarrolló un cuadro sindrómico neurológico con evolución letal.
Tratamiento de la HEMS
El tratamiento de la HEMS es muy discutido. Algunos casos se resuelven espontáneamente, otros con un intenso régimen de transfusiones, radioterapia, cirugía o una combinación de estos tratamientos, según la localización y la presentación.
Comprender los síndromes mielodisplásicos (SMD)
tags: #alteracion #hematopoyesis #en #adulto #mayor