El retraso mental, también conocido como discapacidad intelectual, se caracteriza por limitaciones en el rendimiento que resultan de deficiencias significativas en la inteligencia y en la conducta adaptativa. Esta condición acarrea dificultades clínicas y sociales, generando importantes costos económicos para los sistemas de salud. La comprensión del retraso mental ha evolucionado, reconociendo que la naturaleza y la crianza interactúan en el desarrollo del niño, ampliando la visión tradicional de un enfoque puramente neuropatológico a una evaluación integral de factores biológicos, características familiares adaptativas y el contexto social.
Se define como una función intelectual general por debajo del promedio, con déficit asociado en el comportamiento de adaptación que se manifiesta antes de los 18 años. Su clasificación se basa en el coeficiente intelectual (CI): límite (CI=70-85), ligero (CI=50-69), moderado (CI=35-49), severo (CI=20-34) y profundo (CI=<20).
Síndrome del Cromosoma X Frágil (FRAXA)
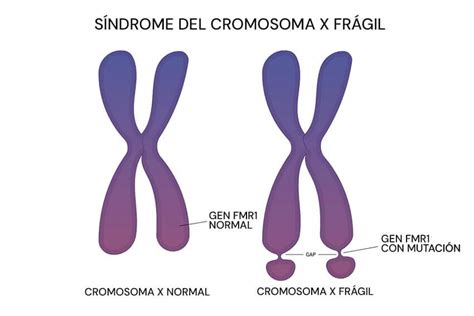
El síndrome del cromosoma X frágil (FRAXA) es la segunda causa genética de retraso mental y la forma más frecuente de retraso mental hereditario. Es una anomalía hereditaria ligada al cromosoma X, responsable de discapacidades que varían desde problemas de aprendizaje hasta retraso mental severo. Este síndrome es causante de la mayoría de los casos de trastornos hereditarios en el desarrollo psicomotor. Se estima una frecuencia de 1:4.000 varones y 1:6.000 mujeres.
Defecto Molecular y Producto Génico
FRAXA obedece a un mecanismo mutacional conocido como mutaciones inestables, amplificación de ADN o expansión de tripletas repetidas. El defecto molecular consiste en una cantidad aumentada de repeticiones de la tripleta CGG (citosina guanina guanina) en una región no codificante del gen FMR1, localizado en el brazo largo del cromosoma X. En la población general, el tracto de repeticiones CGG es polimórfico, con una moda de 29 repeticiones en tándem.
Cuando el número de repeticiones supera aproximadamente las 200, se produce una metilación anormal de una isla CpG, un elemento regulador que controla la transcripción génica. Esta metilación impide la transcripción del gen FMR1 y, por ende, la producción de la proteína FMRP, esencial para el desarrollo neuronal.
La Proteína FMRP
La proteína FMRP, producto del gen FMR1, está ausente en los pacientes con FRAXA. Pesa 69 kDa, se expresa principalmente en el cerebro (específicamente en las neuronas), testículos, placenta y linfocitos. FMRP se une selectivamente a ARNs mensajeros, formando un complejo de ribonucleoproteína mensajera asociado con los polirribosomas. Este complejo es fundamental en las neuronas para regular la traducción de otras proteínas, se localiza en las sinapsis, y su ausencia altera la plasticidad sináptica, que está ligada al aprendizaje y la memoria. Su función es participar en el transporte de ARNs mensajeros desde el núcleo hacia los polirribosomas, principalmente en las dendritas proximales de las neuronas, donde interviene en la traducción de proteínas.
El retraso mental es el resultado de anormalidades en la traducción de proteínas que dependen de FMRP para el transporte de sus ARNs y su traducción. FMRP actúa como represora de la traducción y regula negativamente la traducción de varios ARN específicos de dendritas, algunos de los cuales codifican para proteínas del citoesqueleto y moléculas de transducción de señales, importantes para la plasticidad sináptica. En ausencia de FMRP, se observan niveles aumentados de proteínas relacionadas con funciones neuronales. La inhibición de la traducción a través de FMRP parece ocurrir mediante un mecanismo de interferencia por ARN, un proceso natural para silenciar la expresión génica.
Diagnóstico del Cromosoma X Frágil
Inicialmente, el diagnóstico se realizaba por la expresión citogenética de un sitio frágil en Xq27.3, pero esta prueba ya no se usa debido a su insuficiente sensibilidad y especificidad. Actualmente, se emplean sondas específicas para el diagnóstico directo de la mutación mediante hibridación de Southern, que permite determinar amplificaciones de 80 o más repeticiones. Para diagnosticar portadores de amplificaciones pequeñas, es necesaria la reacción en cadena de la polimerasa (PCR), que permite conocer el tamaño exacto de la región amplificada.
La combinación de Southern y PCR permite discriminar entre:
- Alelo normal: con un máximo de aproximadamente 54 repeticiones de CGG.
- Alelo de portadores asintomáticos: con aproximadamente 55 a 200 repeticiones de CGG.
- Alelo de afectados por la mutación: con más de aproximadamente 200 repeticiones de CGG.
Otra tecnología útil es la secuenciación por fluorescencia láser automática. Estos métodos son exactos, pero costosos, por lo que no se ofrecen a la población general.
Cuadro Clínico del Síndrome de X Frágil
No es posible un diagnóstico clínico definitivo de FRAXA, solo una sospecha que debe confirmarse mediante estudios moleculares. Las alteraciones fenotípicas, enmarcadas dentro del síndrome de Martin-Bell, difieren según el género y suelen aparecer con el crecimiento.
Características Físicas
- Varones antes de la pubertad: Orejas protuberantes, paladar ojival, puente nasal aplastado, macrocefalia, pliegues epicánticos, único pliegue palmar, mala coordinación, articulaciones flexibles e hipotonía.
- Varones después de la pubertad: Cara larga y angosta con mentón prominente, orejas grandes, macro-orquidismo, prolapso de la válvula mitral. También laxitud del tejido conectivo, problemas visuales, escoliosis, tics motores, mayor talla en la niñez y menor en la adultez, piel aterciopelada, pie equino varo, pliegue en la planta del pie, frente ancha o abultada, manos grandes, hernias, pulgares con doble articulación, pie plano y pecho excavado.
- Mujeres: Cara angosta y orejas grandes.
Fenotipo Neurocognitivo
Debido a la inactivación al azar de un cromosoma X, solo el 30-50% de las mujeres portadoras manifiestan síntomas cognitivos. Los individuos sintomáticos (la mayoría de los varones y mujeres afectadas) presentan problemas cognitivos, conductuales y emocionales desde la infancia:
- Retraso cognitivo con coeficientes intelectuales (CI) que disminuyen con la edad.
- Trastornos del lenguaje y la comunicación, con lenguaje expresivo más afectado que el receptivo.
- Desarrollo anormal y reducido de comportamientos de adaptación.
- Anomalías cognitivas en dominios de función ejecutora y cognición visual-espacial.
- Hiperactividad y problemas con sobreestimulación y ansiedad.
Las madres pueden notar atrasos en el desarrollo desde los nueve meses, pero el diagnóstico suele demorarse hasta los tres años por retrasos en el habla o anomalías conductuales. La tasa de desarrollo en preescolares es un tercio o la mitad de lo esperado. El comportamiento autista es evidente desde temprana edad. En la adolescencia, las destrezas adaptativas y cognitivas, así como el funcionamiento de ejecución, declinan en ambos sexos.
Epidemiología
La prevalencia total de FRAXA estimada de estudios moleculares en Inglaterra varía de 1:2.700 a 1:5.700 varones, y es de 1:4.350 en Australia. Un estudio en mujeres de Quebec encontró que 1:500 es portadora de un alelo mayor a 66 repeticiones, y 1:500 es portadora de un alelo entre 55 y 63 repeticiones. Un estudio en la población catalana de España encontró una incidencia de 1 en 2.466 varones afectados y 1 en 411 mujeres portadoras.
El X frágil se ha detectado en todos los grupos étnicos mayoritarios. Se ha observado que un efecto fundador puede ocurrir en algunas poblaciones; por ejemplo, la población judía de Túnez presenta una incidencia diez veces mayor debido a una alta frecuencia de alelos sin la tripleta AGG intercalada.
Prevención y Tratamiento
La prevención primaria de FRAXA se basa en la asesoría genética oportuna a los miembros de la familia afectada, siendo la forma más efectiva de enfrentar este síndrome. En Nueva Gales del Sur, Australia, la tasa de incidencia ha disminuido diez veces (de 4.3/10.000 a 0.5/10.000) gracias a un programa de consejo genético, diagnóstico prenatal e interrupción del embarazo.
La prevención secundaria (post-concepción) se logra en países que permiten la interrupción del embarazo mediante diagnóstico precoz (embrionario o fetal). En cuanto a la prevención terciaria, el estudio profundo de los pacientes ha permitido plantear estrategias específicas para mejorar su funcionamiento intelectual y adaptación social, de ahí la importancia de la confirmación molecular del diagnóstico clínico.
El tratamiento del síndrome del cromosoma X frágil se enfoca en estrategias para mejorar el funcionamiento intelectual y la adaptación social.
Retraso Mental de Causa Genética en Rovira (Colombia)
En Colombia, la mayoría de los pacientes con retraso mental no reciben evaluación genética. Un estudio preliminar en Rovira (Tolima), una población de aproximadamente 24.162 habitantes, identificó una frecuencia aproximada de 1/300 casos de discapacidad, aparentemente por retraso mental. Se realizó un diagnóstico clínico preliminar en 25 pacientes, junto con pruebas bioquímicas y citogenéticas (Bandeo G).
Se identificó una familia con tres hijos afectados (dos mujeres y un hombre) con retraso mental y braquidactilia, lo que sugiere un componente genético. Las pruebas metabólicas fueron negativas y los cariotipos normales. Se plantea la necesidad de realizar pruebas moleculares, incluyendo el síndrome de X-frágil, para complementar el estudio y brindar consejería genética.
Factores Ambientales en Rovira
La economía de Rovira es principalmente agrícola, basada en cultivos como sorgo, arroz, algodón, café y tomate. También hay actividad minera (carbón, hierro, mercurio, cobre, oro, plomo, zinc), lo que puede implicar la presencia de compuestos genotóxicos. Un muestreo aleatorio de agua y tomate en Rovira para detectar plaguicidas resultó 100% positivo para organofosforados en todas las muestras. Para carbamatos, se reportó un 60% de positividad en muestras de agua y 100% en muestras de tomate. Todas las muestras fueron negativas para organoclorados. No obstante, el estudio no buscaba asociar directamente los plaguicidas con el retraso mental, sino identificar posibles factores de riesgo.
Prevalencia y Etiología de la Discapacidad Intelectual a Nivel Global
La prevalencia de la discapacidad intelectual (DI) se estima entre 1% y 4% a nivel mundial. La Organización Mundial de la Salud (OMS) estima un 10% de personas con DI en países del tercer mundo. Se han identificado más de 250 causas de DI, cuya etiología es múltiple.
Un estudio realizado en Cuba, y posteriormente en otros cinco países, investigó a 1.207.472 personas con discapacidades en una población general de 64.786.057 habitantes, arrojando una tasa de prevalencia de 1.86 por cada 100 habitantes de todas las edades.
Estudio en Angola
En Angola, un estudio observacional descriptivo transversal prospectivo evaluó a 139 escolares con discapacidad intelectual. La etiología prenatal fue la más frecuente (45,3%), seguida de la posnatal (29,5%) y perinatal (18,0%).
- Etiología prenatal: Predominaron los factores ambientales (20,9%), destacando el consumo de alcohol (8,6%). Al profundizar, predominaron las enfermedades monogénicas. El alcoholismo se manifestó con signos dismórficos en el 100% de los casos de síndrome alcohólico fetal. También se identificaron 13 casos con síndromes genéticos, 4 con aberraciones cromosómicas y 6 con trastorno multifactorial debido a agregación familiar.
- Etiología perinatal: La hipoxia al nacer (5,0%) fue la causa más frecuente, seguida de infecciones, traumatismos y convulsiones.
- Etiología posnatal: La malaria cerebral (15,1%) fue la causa principal y la tercera más predominante en general.
El 80% de los casos estudiados tenían al menos un familiar con la misma discapacidad. Los antecedentes maternos predominaron (51,4%) sobre los paternos (21,9%). El 37,5% de los casos tenían familiares de primer grado afectados, destacando la madre con DI (30,1%).
Finalmente, el 25,1% de todos los casos analizados tenían una causa genética, el 6,5% era indeterminado y el 68,4% tenía una causa ambiental. Estos hallazgos sugieren la necesidad de implementar una estrategia de atención prenatal, perinatal y posnatal en Angola, además de servicios de asesoramiento genético y control de infecciones como la malaria.
Retraso Mental de Causa Genética en Atención Primaria (España)
Un estudio observacional en un centro de atención primaria en Zaragoza (España) revisó los trastornos del neurodesarrollo en la población pediátrica. De 76 casos de trastornos del neurodesarrollo (2,19% de la población), 11 correspondieron a retraso mental. Se solicitó estudio genético en 21 casos (27,63%), siendo más frecuente en casos de retraso mental o Trastorno del Espectro Autista (TEA).
Las pruebas genéticas proporcionaron un diagnóstico en 12 pacientes (57,14% de los estudiados). En general, se encontró una causa genética en el 75% de los casos con diagnóstico establecido. Este estudio subraya la importancia creciente del diagnóstico genético preciso en trastornos del neurodesarrollo.
Retraso Mental como Secuela de Enfermedad Neurometabólica
Panorama de tratamiento de las enfermedades neurometabólicas en la última década
Los Errores Innatos del Metabolismo (EIM) son defectos en la actividad de proteínas esenciales para el metabolismo celular, representando un grupo paradigmático de enfermedades raras. Actualmente, se han caracterizado más de 500 EIM gracias a los avances en genética, biología molecular y bioquímica. La mayoría de los EIM presentan un patrón de herencia autosómico-recesivo, con algunos casos de herencia autosómica-dominante o ligada al cromosoma X. Estos defectos pueden generar depósito de sustrato, déficit de producto o activación de vías metabólicas alternativas que producen metabolitos tóxicos.
El conocimiento de las bases moleculares ha mejorado las posibilidades de diagnóstico prenatal y neonatal, permitiendo la implementación de programas de manejo preventivo, tratamiento precoz y consejo genético oportuno. Sin embargo, un gran número de casos permanecen sin diagnóstico, dificultando el enfoque terapéutico completo.
Caso Clínico y Diagnóstico
Se reporta el caso de un adolescente de 17 años con retraso mental y epilepsia mioclónica progresiva degenerativa. Presentó movimientos tónico-clónicos desde los 9 meses hasta los 2 años, desencadenados por un episodio febril, y un retraso del desarrollo psicomotor hacia el primer año de vida. A los ocho años, sufrió otra crisis tónico-clónica generalizada.
En la valoración genética, se encontraron anomalías fenotípicas como macrocefalia e hipertelorismo, genu varo izquierdo, pie plano con hallux valgo bilateral, clinodactilia, cifoescoliosis e hipotonía troncular. El electroencefalograma fue severamente anormal y la resonancia magnética cerebral mostró leucodistrofia, ensanchamiento de las cisuras de Silvio, pérdida de volumen fronto-temporal y ectasia leve del sistema ventricular. Un SPECT cerebral evidenció áreas fronto-temporales con deficiente irrigación.
Se planteó la hipótesis diagnóstica de EIM. Las pruebas bioquímicas revelaron niveles de amonio en plasma por encima de lo normal (más de 50 Umol/L, referencia 9-30 Umol/L), niveles de ácido glutámico hasta 10 veces por encima de lo normal, y alteraciones en glicina y alanina. Los estudios para daño enzimático mitocondrial específico en la vía de la carnitina estuvieron dentro del rango. El manejo terapéutico inicial incluyó benzoato de sodio, L-carnitina, coenzima Q, arginina, clobazam, levetiracetam, vigabatrin y fenobarbital, con respuesta favorable.
Aciduria Glutárica Tipo 1 (AcGLUT-1)
Después de un análisis de la historia clínica, se consideró la Aciduria Glutárica Tipo 1 (AcGLUT-1) como uno de los diagnósticos diferenciales más importantes. Esta patología es causada por la deficiencia de la enzima Glutaril Co-A deshidrogenasa, fundamental para el metabolismo de lisina, hidroxilisina y triptófano. El déficit enzimático lleva a la acumulación de sustancias como ácido glutárico, ácido 3-hidroxiglutárico, ácido glutacónico y glutarilcarnitina.
AcGLUT-1 es de herencia autosómica recesiva, con el gen localizado en el cromosoma 19p13.2. Su prevalencia global es de 1 en 100.000 recién nacidos. Clínicamente, se caracteriza por macrocefalia y un periodo asintomático seguido de eventos de hipercatabolismo (ej. cuadro febril leve) que desencadenan sintomatología extrapiramidal. La acumulación de ácido glutámico causa neurotoxicidad en la corteza frontotemporal y ganglios basales, resultando en crisis epilépticas, incoordinación motora, alteraciones del lenguaje e hipotonía.