El Síndrome de Williams, también conocido como síndrome de Williams-Beuren (SWB), es un raro trastorno genético neurodegenerativo que afecta múltiples sistemas del cuerpo. Se caracteriza por un grado variable de discapacidad intelectual, un perfil de personalidad con rasgos únicos, distintivas características faciales y problemas cardíacos y vasculares. Este síndrome afecta por igual a hombres y mujeres, con una prevalencia estimada de uno de cada 7.500 a 20.000 nacidos vivos a nivel mundial, sin predilección por una zona geográfica específica.

Causas Genéticas del Síndrome de Williams
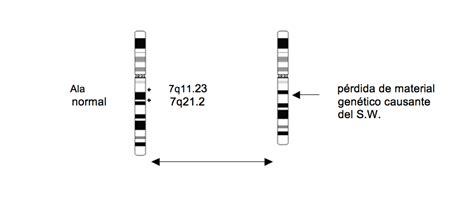
El Síndrome de Williams es causado por la pérdida, o deleción, de un segmento de material genético en una región específica del cromosoma 7. Esta deleción ocurre en el brazo largo (q) del cromosoma 7, específicamente en la región de las bandas 11.23 (7q11.23), y abarca aproximadamente entre 25 y 28 genes contiguos. Los cromosomas, que son los portadores de la información genética de cada individuo, están presentes en el núcleo de las células humanas y se subdividen en bandas que se numeran, permitiendo la localización precisa de esta anomalía.
Los investigadores creen que la pérdida de varios de estos genes es la responsable de las características del síndrome. Entre los genes que suelen estar deletados se incluyen CLIP2, ELN, GTF2I, GTF2IRD1, y LIMK1:
- El gen ELN (elastina) se ha vinculado específicamente con las anomalías del tejido conectivo y los problemas cardiovasculares, como la estenosis aórtica supravalvular.
- Las deleciones de los genes CLIP2, GTF2I, GTF2IRD1 y LIMK1 parecen estar implicadas en las dificultades características con las tareas visoespaciales, el comportamiento peculiar y otras dificultades cognitivas observadas en estas personas. Se cree que el gen LIMK1 está particularmente involucrado en los problemas visoespaciales.
- La presencia o ausencia del gen NCF1, también localizado en el cromosoma 7, influye en el riesgo de desarrollar hipertensión arterial en personas con Síndrome de Williams. Cuando este gen está deletado, hay menos probabilidades de desarrollar hipertensión, lo que sugiere que su pérdida puede ser un factor protector.
La mayoría de los casos del Síndrome de Williams no se heredan, sino que ocurren de manera esporádica (de novo) como un evento aleatorio durante la formación de las células reproductivas (óvulos o espermatozoides) en uno de los padres. Sin embargo, el síndrome se considera un trastorno autosómico dominante, lo que significa que una persona afectada tiene un 50% de posibilidades de transmitir la deleción a su descendencia. En casos en que los padres no presentan síntomas, el riesgo para los hermanos de un afectado es bajo.
Características Clínicas Distintivas
El Síndrome de Williams se manifiesta a través de un amplio espectro de síntomas y características físicas que varían en alcance y gravedad entre los individuos. Las personas con el síndrome no presentan necesariamente todos los síntomas enumerados.
Rasgos Físicos
Los niños pequeños con Síndrome de Williams a menudo muestran rasgos faciales distintivos, conocidos como "cara de elfo", que pueden incluir:
- Frente amplia y prominente.
- Nariz pequeña, con un puente nasal aplanado y fosas nasales que se voltean hacia arriba (narinas antivertidas) o una punta ancha y respingada.
- Mejillas llenas y labios gruesos, con una boca grande que generalmente se mantiene abierta.
- Ojos grandes, a menudo con pliegues palpebrales cortos ("ojos achinados").
- Cejas anchas.
- Mandíbula inferior pequeña y orejas grandes.
- Problemas dentales como dientes pequeños, muy espaciados o torcidos, esmalte dental defectuoso o dientes ausentes.
Otros rasgos físicos pueden incluir baja estatura y un tórax hundido, además de problemas en las articulaciones y un bajo tono muscular.
Perfil Cognitivo y Conductual
Un aspecto central del Síndrome de Williams es la presencia de un retraso en el desarrollo, que puede manifestarse como una discapacidad intelectual leve a moderada. Los niños suelen mostrar retrasos psicomotores en los primeros meses de vida, por ejemplo, para empezar a caminar o hablar.
A pesar de la discapacidad intelectual, un rasgo distintivo es el perfil cognitivo particular:
- Fortalezas lingüísticas: Tienen buenas habilidades verbales y del lenguaje, a menudo con una tendencia a la locuacidad, y una fuerte capacidad para aprender escuchando.
- Déficits visoespaciales: Presentan dificultades con las tareas visuales y espaciales y con los números.
- Memoria a corto plazo: Suelen tener una buena memoria a corto plazo.
El perfil conductual y social es notablemente único. Las personas con Síndrome de Williams son generalmente muy amigables, extrovertidas, empáticas y tienen una gran voluntad de agradar. Son los primeros en saludar, participar en charlas y son descritos como hipersociables. Sin embargo, esta sociabilidad excesiva puede ser paradójica:
- Son muy confiados con los desconocidos y no suelen entender cuándo están en peligro o son víctimas de engaños, lo que los hace muy vulnerables.
- Aunque pueden entablar conversaciones iniciales sin problemas, a menudo carecen de buenas habilidades sociales para mantener relaciones profundas o duraderas con sus iguales a largo plazo, lo que puede llevar al aislamiento social y la soledad en la adultez.
- Pueden tener dificultades para captar adecuadamente las normas sociales o respetar los turnos en el uso de la palabra, lo que dificulta la fluidez comunicativa.
- La desconexión entre su cordialidad extrema y su incapacidad para entender las reacciones de los demás puede generarles ansiedad.
- También pueden presentar tendencia a la distracción y trastorno de hiperactividad con déficit de atención (THDA).
Problemas Cardiovasculares y Otros Síntomas
Los problemas cardiovasculares son muy frecuentes y a menudo el problema médico más grave asociado al síndrome. Incluyen:
- Estenosis aórtica supravalvular: Un estrechamiento de la aorta, la arteria principal que lleva sangre desde el corazón al resto del cuerpo. Si no se trata, puede causar dificultad para respirar, dolor en el pecho e insuficiencia cardíaca.
- Estenosis de las arterias pulmonares: Estrechamiento de las arterias que van a los pulmones.
- Hipertensión arterial.
- Problemas en las válvulas aórtica y mitral del corazón.
Otras anomalías y síntomas comunes incluyen:
- Hipercalcemia infantil: Niveles anormalmente elevados de calcio en la sangre durante la infancia, que pueden causar cólicos, irritabilidad, vómitos, convulsiones y rigidez muscular.
- Hipersensibilidad auditiva (hiperacusia): Muchos sonidos cotidianos (como una aspiradora) pueden resultarles desagradables o incluso dolorosos.
- Afinidad por la música: A pesar de la hipersensibilidad, muchas personas con Síndrome de Williams tienen una sensibilidad especial e innata para la música, y hay más casos de oído absoluto en esta población. Algunos pueden desarrollar talento musical formal.
- Problemas musculoesqueléticos: Bajo tono muscular, problemas en las articulaciones y cambios en las curvaturas de la columna vertebral (lordosis, cifosis y escoliosis). Las articulaciones débiles pueden volverse rígidas con la edad.
- Problemas renales y del tracto urinario: Anomalías de los riñones, infecciones crónicas del tracto urinario, conducto lagrimal obstruido.
- Problemas digestivos: Irritabilidad, cólicos, reflujo, vómitos y estreñimiento.
- Problemas endocrinos: Hipotiroidismo, obesidad y una mayor frecuencia de diabetes.
- Problemas visuales: Hipermetropía y estrabismo.
Los Reporteros | ¿Qué es el síndrome de Williams?
Diagnóstico del Síndrome de Williams
El Síndrome de Williams fue identificado por primera vez en 1961. Los primeros meses de vida hasta el diagnóstico suelen ser difíciles para las familias debido a la incertidumbre, ya que algunos síntomas tempranos como la irritabilidad y problemas digestivos pueden confundirse con características normales de los recién nacidos. Aunque el aspecto del recién nacido puede generar sospechas, en algunos casos los rasgos faciales no son evidentes al nacer. Los médicos suelen considerar la posibilidad de Síndrome de Williams cuando hay retrasos en el desarrollo u otros síntomas alrededor de los dos años de edad.
El diagnóstico puede confirmarse mediante una evaluación clínica detallada, incluyendo un historial completo del paciente, y análisis especializados. Las pruebas genéticas son fundamentales para la confirmación:
- Hibridación fluorescente in situ (FISH): Es la forma más clásica de determinación y se utiliza para detectar la deleción del gen de la elastina (ELN) en el cromosoma 7. Esta técnica de laboratorio utiliza una sonda de ADN marcada con una sustancia fluorescente que se une a la secuencia de ADN correspondiente en el cromosoma, permitiendo visualizar la ausencia de los genes en la región q11.23 del cromosoma 7 bajo un microscopio especial.
- Array CGH (hibridación genómica comparada en microarrays): Más recientemente, esta técnica permite detectar deleciones menores y atípicas que FISH no reconoce. Consiste en comparar el ADN del paciente con el de una persona no portadora, utilizando fluorescencia para detectar pérdidas genéticas.
Hoy en día, en países como España o Reino Unido, el diagnóstico tiende a realizarse durante el primer año de vida, gracias a un mayor conocimiento y a que las señales de alarma "saltan enseguida". Sin embargo, para personas con Síndrome de Williams de 20 o 30 años, los diagnósticos llegaron en la segunda infancia o incluso en la adolescencia. Las personas mayores de 35 años con el síndrome a menudo no están identificadas como tal y son tratadas simplemente como personas con discapacidad intelectual sin un diagnóstico específico que explique sus dificultades.
Tratamiento y Manejo del Síndrome de Williams
Dado que las enfermedades de origen genético no tienen cura, el tratamiento del Síndrome de Williams se centra en reducir el efecto de los síntomas, mejorar la calidad de vida y prevenir complicaciones. Para ello, se requiere un enfoque multidisciplinario que involucre a diversos especialistas a lo largo de la vida del individuo.
Intervenciones Específicas por Síntoma:
- Problemas cardíacos: Es fundamental una evaluación minuciosa por cardiología tras el diagnóstico, con un seguimiento frecuente al menos hasta los 4 años. Algunas estenosis pueden requerir cirugía, mientras que otras, más raramente, pueden mejorar con medicamentos, fisioterapia y ejercicio específico, dependiendo de la ubicación y gravedad.
- Hipercalcemia: Para bebés con niveles elevados de calcio, se puede prescribir una dieta que restrinja la ingesta de vitamina D y calcio. En casos graves, se puede considerar el tratamiento temporal con un corticoide como la prednisona. Después de los 12 meses de edad, los niveles de calcio suelen normalizarse, incluso sin tratamiento.
- Hipersensibilidad auditiva: Puede tratarse con protección auditiva cuando se pueden predecir aumentos de los niveles de ruido. Se recomienda la consulta con un otorrinolaringólogo para infecciones recurrentes del oído (otitis media), que pueden requerir tubos de timpanotomía.
- Desarrollo y aprendizaje: Los centros para niños con discapacidades del desarrollo y los servicios de educación especial son beneficiosos. Un equipo de apoyo que incluya terapia del habla y del lenguaje, fisioterapia, terapia ocupacional, servicios sociales y/o capacitación vocacional es crucial para que alcancen su potencial personal.
- Problemas oculares: La hipermetropía se trata con lentes correctivos. Se recomienda la consulta con un oftalmólogo.
- Cuidado dental: Puede requerir asistencia con el cepillado diario y el uso de hilo dental, con limpiezas dentales cada 4 meses en adolescentes y adultos.
- Estreñimiento: Generalmente incluye aumentar la ingesta de agua y fibra, seguido de laxantes osmóticos si es necesario.
- Rigidez articular: La fisioterapia es fundamental para manejar este síntoma.
Es importante que los niños reciban atención en clínicas especializadas en Síndrome de Williams siempre que sea posible. El asesoramiento genético es altamente recomendado para las personas afectadas y sus familias.
Pronóstico y Calidad de Vida
La expectativa de vida de las personas con Síndrome de Williams ha mejorado notablemente en los últimos tiempos, ubicándose alrededor de los 52 años, siempre y cuando se monitoreen y traten adecuadamente los problemas de salud mayores, especialmente los cardíacos, que son mucho más frecuentes que en la población general. Las personas con Síndrome de Williams suelen tener un cierto nivel de discapacidad intelectual y, en la mayoría de los casos, requieren cuidadores a tiempo completo y a menudo viven bajo supervisión, incluso en la edad adulta.
Aunque no hay cura, los tratamientos y terapias pueden mitigar muchos de los problemas. Obtener ayuda desde el primer momento es crucial para que los niños alcancen su máximo potencial. Conectarse con otras familias y grupos de apoyo también puede ser de gran ayuda, proporcionando información valiosa y recursos. Es esencial una planificación nutricional para abordar problemas como la obesidad y la diabetes.
