El síndrome de West (SW), también conocido como espasmos infantiles, es una encefalopatía epiléptica pediátrica grave y rara, dependiente de la edad. Se caracteriza por una tríada electroclínica distintiva que incluye espasmos epilépticos, un retardo o detención del desarrollo psicomotor, y un patrón electroencefalográfico de hipsarritmia, aunque uno de estos elementos puede estar ausente. Esta condición se inicia en la mayoría de los pacientes durante el primer año de vida, generalmente entre los 3 y 12 meses, con un pico entre los 4 y 6 meses de edad.
El término de encefalopatía epiléptica se refiere a una condición en la cual se piensa que las anomalías epileptiformes contribuyen al disturbio progresivo en la función cerebral. Originalmente, los espasmos infantiles fueron descritos por West en 1841, quien los observó en su propio hijo de 4 meses. Cerca de un siglo después, en 1952, Gibbs y Gibbs describieron el patrón electroencefalográfico de hipsarritmia, caracterizado por "puntas y ondas lentas de gran amplitud, desordenadas, que varían de un momento a otro tanto en duración como en localización". A partir de la década de 1960, la combinación de espasmos infantiles, retardo psicomotor y hipsarritmia fue denominada síndrome de West.
Epidemiología e Incidencia
El síndrome de West representa la forma más frecuente de epilepsia en el primer año de vida, excluyendo las convulsiones neonatales y crisis febriles. La incidencia de este síndrome varía en diferentes estudios, oscilando entre 1 por 4.000 a 6.000 nacidos vivos, o alrededor de 1.6 a 2.0 por cada 10.000 niños menores de 10 años. En algunas series se ha reportado un predominio en el sexo masculino, aunque otros estudios no han encontrado una significación estadística en esta diferencia. La incidencia familiar es baja, excepto en subgrupos con características genéticas dominantes como la esclerosis tuberosa, y los antecedentes familiares de otras epilepsias son poco frecuentes.
Características Clínicas
La Tríada Clásica
La tríada que define el síndrome de West incluye:
- Espasmos epilépticos: Son las crisis características, que pueden ser en flexión, extensión o mixtos. Consisten en contracciones de carácter axial, bruscas y breves, que pueden ir acompañadas de llanto, contracciones del cuello y nistagmo. A través de los años, estas crisis han sido denominadas espasmos infantiles, pero la Liga Internacional contra la Epilepsia propuso sustituir este término por espasmos epilépticos.
- Retardo del desarrollo psicomotor: Uno de los principales efectos, y un componente crucial del síndrome. Esto puede afectar la capacidad del bebé para adquirir hitos como sentarse, gatear, caminar o hablar. En muchos casos, los pacientes experimentan una regresión del neurodesarrollo.
- Hipsarritmia en el electroencefalograma (EEG): Un patrón electroencefalográfico desorganizado por ondas lentas bilaterales, simétricas y sin sincronía interhemisférica, con trazado caótico de alto voltaje y localización difusa.
Otros Síntomas y Consecuencias
Además de la tríada clásica, los niños con síndrome de West pueden presentar otros síntomas y consecuencias:
- Pérdida del sentido del humor e indiferencia ante estímulos sensoriales.
- Mayor irritabilidad y llantos sin motivo aparente.
- Dificultad para conciliar el sueño y pérdida de horas de descanso.
- Las crisis epilépticas frecuentes pueden poner en riesgo el bienestar del bebé y afectar su calidad de vida.

Clasificación Etiológica
Según la Clasificación Internacional de las Epilepsias y síndromes epilépticos, el síndrome de West se clasifica según su etiología en sintomático, criptogénico e idiopático. Recientemente, el grupo de trabajo para la Clasificación y Terminología de la Liga Internacional contra la Epilepsia propuso sustituir el término criptogénico por “probablemente sintomático” para reflejar la presunción de una causa subyacente.
Grupo Sintomático
Constituye el grupo más frecuente, representando entre el 45.7% y el 67% de los casos en diversas series. Se refiere a un síndrome en el cual las crisis son el resultado de una o más lesiones estructurales cerebrales identificables. Las causas se agrupan en factores etiológicos prenatales, perinatales y posnatales:
- Causas Prenatales: Son las más frecuentes, constituyendo entre el 22.3% y el 42.8% de los pacientes. El surgimiento de técnicas de neuroimagen avanzadas como la tomografía axial computarizada (TAC), la resonancia magnética nuclear (RMN), la tomografía por emisión de positrón (PET) y la tomografía computarizada por emisión de positrón único (SPECT) ha contribuido significativamente al diagnóstico de estas causas.
- Displasias Cerebrales: Displasia cortical (la etiología más frecuente, alrededor del 30%), esclerosis tuberosa, neurofibromatosis, incontinencia pigmenti, síndrome de Sturge-Weber, síndrome del nevus linear sebáceo, hemangiomatosis neonatal, síndrome del nevus epidérmico con hemimegalencefalia, síndrome de Aicardi, lisencefalia, hemimegalencefalia, displasia focal cortical, paquigiria, heterotopias, holoprosencefalia, esquizencefalia, displasia septo-óptica, tuberosidades solitarias corticales, agenesia o disgenesia del cuerpo calloso, agenesia septal, microcefalia congénita.
- Anomalías Cromosómicas: Síndrome de Down (la trisomía 21 es una causa importante), síndrome de Miller Dieker, trisomía 7q, trisomía parcial 2p, duplicación 18q, tetrasomía 15p, duplicación 15q, monosomía 18p.
- Infecciones: Citomegalovirus, herpes simple, rubéola, toxoplasmosis, sífilis.
- Enfermedades Metabólicas: Fenilcetonuria, hiperglicinemia no cetósica, hiperornitinemia, homocitrulinemia, síndrome de Leigh, deficiencia de piruvato carboxilasa, deficiencia de piruvato deshidrogenasa, dependencia de piridoxina, enfermedad de Krabbe, adrenoleucodistrofia neonatal, leucodistrofia ortocromática, encefalopatía por glicina, deficiencia de biotinidasa, deficiencia del complejo I de la cadena respiratoria, deficiencia de nitocromo c oxidasa.
- Síndromes Congénitos: Síndrome de Sjogren-Larsson, síndrome de CHARGE, síndrome de PEHO, síndrome de Smith-Lemli-Optiz, enfermedad de Fahr, entre otros.
- Insulto Hipóxico-Isquémico: Poroencefalia, hidranencefalia, leucomalacia periventricular.
- Causas Perinatales: Incluyen lesiones difusas a causa de encefalopatía hipóxico-isquémica y lesiones focales por trastornos cerebrovasculares (entre las 28 semanas de embarazo y los primeros 7 días de vida). Después de las causas prenatales, son las más frecuentes, identificándose en el 13.9% al 22% de los pacientes. Ejemplos incluyen necrosis selectiva neural, status marmoratus, daño cerebral parasagital, leucomalacia periventricular, necrosis isquémica focal y multifocal (poroencefalia, encefalomalacia multiquística), e hipoglicemia.
- Causas Posnatales: Constituyen las causas menos frecuentes del síndrome de West, encontrándose en el 6.7% al 8.5% de los casos. Predominan las infecciones (meningitis bacteriana o viral, absceso cerebral), hemorragias y traumas (subdural, subaracnoidea), encefalopatía hipóxico-isquémica (paro cardíaco), y tumores cerebrales. No se ha demostrado que la vacunación contra la difteria-tétanos-tos ferina (DPT) sea una causa, sugiriéndose que su asociación es una coincidencia debido a la edad de administración.

Grupo Criptogénico
Se refiere a aquellos síndromes en los cuales se presume una causa sintomática, pero esta permanece oculta tras un estudio etiológico exhaustivo. La proporción de síndrome de West criptogénico varía ampliamente entre investigadores (del 9% al 41%), dependiendo de las definiciones utilizadas y el momento del diagnóstico. Este grupo se caracteriza por un desarrollo psicomotor normal al inicio de los espasmos, que en la mayoría de los casos se deteriora progresivamente.
Grupo Idiopático
Aunque la Clasificación Internacional de las Epilepsias y síndromes epilépticos no reconoce formalmente la etiología idiopática del síndrome de West, varios autores han reportado su existencia. Incluye casos en los que no existe una lesión estructural subyacente ni anomalías neurológicas. Estos casos suelen tener una evolución favorable con desaparición de las crisis y un desarrollo psicomotor normal. Las características fundamentales de estos niños incluyen la ausencia de regresión mental significativa, preservación de la función visual, ausencia de anomalías electroencefalográficas interictales focales y la presencia de espasmos simétricos con reaparición de la hipsarritmia entre espasmos consecutivos de una salva. Puede existir una predisposición genética o antecedentes familiares de otras formas de epilepsia idiopática o crisis febriles.
Fisiopatología
La fisiopatología del síndrome de West es aún desconocida, pero se han postulado diversas hipótesis. Se cree que los espasmos constituyen una respuesta inespecífica de un cerebro inmaduro a cualquier daño. La edad de inicio del síndrome coincide con el período crítico de formación de dendritas y mielinización, lo que podría contribuir a la fisiopatología.
Una hipótesis sugiere que un desequilibrio de los neurotransmisores del tallo cerebral podría ser responsable de los espasmos y la hipsarritmia, ya sea por un incremento de la actividad de los sistemas adrenérgicos y/o serotoninérgicos, o por una disminución de la actividad del sistema colinérgico. Esto se apoya en la disminución de la duración del sueño REM en estos pacientes y la observación de la desaparición de los espasmos y reducción de la hipsarritmia durante esta etapa del sueño. En pacientes con espasmos epilépticos y síndrome de Down, se han planteado anomalías en la función del receptor glutamato, con niveles elevados de este neurotransmisor.
Estudios de metabolismo cerebral mediante PET apoyan la participación de estructuras subcorticales (núcleo lenticulado y tallo cerebral) en el origen de los espasmos y la hipsarritmia. Asimismo, diversas lesiones corticales como malformaciones cerebrales y síndromes neurocutáneos, sugieren la participación de la corteza cerebral. Se ha demostrado que las descargas corticales focales pueden preceder o asociarse con los espasmos.
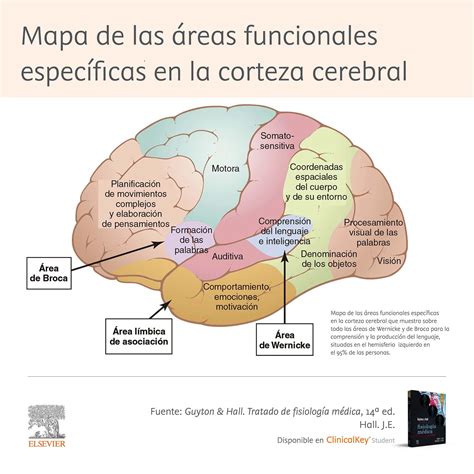
Retraso del Desarrollo y Comorbilidades
Impacto en el Neurodesarrollo
El retraso del desarrollo neurológico es uno de los principales efectos del síndrome de West, afectando significativamente las habilidades psicomotoras y cognitivas del niño. Tanto las crisis epilépticas como los tratamientos antiepilépticos pueden favorecer déficits cognitivos variables. Esto ha provocado un aumento del interés por una temprana evaluación neuropsicológica adaptada a esta población.
Comorbilidad con el Trastorno del Espectro del Autismo (TEA)
El síndrome de West se ha asociado con el Trastorno del Espectro del Autismo (TEA), uno de los trastornos más importantes del neurodesarrollo. La gran heterogeneidad de síntomas que caracterizan a ambos cuadros hace que esta asociación no sea sorprendente. Aproximadamente el 80% de los pacientes con epilepsia o TEA presentan discapacidad intelectual, lo que podría explicar por qué la epilepsia se asocia más con la discapacidad intelectual que con el autismo. Sin embargo, dado que pacientes con TEA con discapacidad leve o habilidades excepcionales (Savant) también presentan crisis epilépticas, se considera adecuado hablar de comorbilidad.
Los principales criterios diagnósticos del DSM-5 para el TEA incluyen dificultades en la interacción social y comunicación, retraso y/o alteración en el lenguaje, y la presencia de patrones estereotipados, comportamiento restrictivo e intereses particulares.
Alteraciones Cognitivas y Emocionales
Las alteraciones cognitivas y emocionales en niños con síndrome de West y TEA asociado no distan en exceso de las secuelas de la epilepsia por sí misma, aunque la comorbilidad de ambos síndromes podría aumentar la gravedad de dichas alteraciones. A nivel cognitivo, esta población presenta alteraciones en:
- Memoria
- Aprendizaje
- Atención
- Visopercepción
- Lenguaje
- Funciones ejecutivas (las alteraciones en estas funciones se han asociado con una pobre cognición social).
Las alteraciones neuropsicológicas y emocionales habituales en pacientes con síndrome de West y TEA asociado producen un empeoramiento de su calidad de vida. Por ello, el foco terapéutico debe centrarse principalmente en la mejora de la autonomía y la calidad de vida del paciente.
Diagnóstico
Es fundamental diagnosticar el síndrome de West lo antes posible para evitar el deterioro cognitivo y secuelas que aumenten la morbimortalidad de los pacientes. La neuroimagen es la prueba con mayor rentabilidad diagnóstica; aproximadamente el 70% de los pacientes alcanzan la causa subyacente tras una correcta anamnesis, examen físico y la realización de una resonancia magnética craneal. Trastornos de la migración neuronal y de la mielinización pueden demostrarse mejor mediante la resonancia magnética nuclear. La tomografía axial computarizada puede diagnosticar malformaciones mayores o disgenesias.
En niños sin diagnóstico etiológico tras el estudio inicial, un estudio coste-eficiente incluiría la realización de un CGHa (Array Híbrido Comparativo Genómico), seguido de un panel de epilepsia y un estudio metabólico general (ácido láctico, ácido pirúvico, aminoácidos y ácidos orgánicos en orina). Estos estudios pueden alcanzar el diagnóstico etiológico en un 10-15% adicional de casos.
Pronóstico
Variabilidad del Pronóstico
El pronóstico para los niños con síndrome de West varía considerablemente. Algunos pueden lograr una mejora significativa con el tratamiento, mientras que otros pueden enfrentar discapacidades a largo plazo. La muerte prematura ocurre en el 5% al 31% de los casos. El pronóstico global del SW criptogénico ha resultado ser más grave de lo esperado en algunos estudios, con una evolución desfavorable en una proporción significativa de pacientes.
A pesar de los avances en el conocimiento del síndrome, aún hay poca especialización en este campo, y algunos casos llegan a ser detectados tardíamente debido a malinterpretaciones del pediatra.
Factores Pronósticos
Se han estudiado diversas variables en busca de factores que interfieran en el pronóstico:
- El sexo femenino, el comienzo tardío y el control precoz de la hipsarritmia han resultado ser factores de buen pronóstico en algunos estudios.
- Una mayor evolución hacia otras formas de epilepsia se ha observado en el sexo masculino y en los casos con inicio antes de los 6 meses.
- El tratamiento temprano (en las primeras 2 semanas del comienzo de los espasmos) y el control precoz de la hipsarritmia (en las primeras 3 semanas) se han asociado con una menor evolución hacia epilepsia secuelar.
Evolución a Otras Epilepsias
Un porcentaje de pacientes puede evolucionar a otras formas de epilepsia. Por ejemplo, en un estudio, el 44% de los pacientes evolucionaron a otras epilepsias, apareciendo en los primeros 2 años del diagnóstico. Aunque la incidencia del síndrome de Lennox-Gastaut fue baja, la epilepsia focal resultó ser la evolución más frecuente.
Un caso clínico ilustrativo es el de un lactante de 5 meses que presentó irritabilidad, dificultad para conciliar el sueño, desviación de la mirada y movimientos tónicos del hombro. Se le realizó un electroencefalograma, observándose el patrón de hipsarritmia, y se inició tratamiento con ácido valproico. A los 10 años, el paciente presentó una severa escoliosis toraco-lumbar, múltiples hospitalizaciones por neumonía, y se le realizó gastrostomía debido a una complicación, falleciendo a los 13 años por neumonía grave. Este caso resalta la gravedad y el impacto multifactorial del síndrome.
Tratamiento y Programas de Intervención
El tratamiento del síndrome de West tiene como objetivo principal disminuir las convulsiones con la mínima dependencia necesaria de medicamentos. Es crucial un manejo integral que abarque tanto los aspectos farmacológicos como las intervenciones neuropsicológicas.
Tratamiento Farmacológico y Quirúrgico
Las principales terapias que han demostrado ser eficaces son la vigabatrina (VGB) y la hormona adrenocorticotrópica (ACTH). En algunos estudios, el tratamiento con ACTH ha producido un mayor efecto sobre el control de los espasmos a corto plazo, aunque no se ha demostrado su superioridad a largo plazo. Lamentablemente, ningún tratamiento médico proporciona un alivio satisfactorio a todos los infantes. Sin embargo, en algunos casos, la resección cortical de áreas específicas en el sistema nervioso central puede llevar a una disminución notable de las convulsiones.
Se han reportado efectos secundarios con ambos tratamientos, siendo más frecuentes y severos con la ACTH (hipertensión arterial, hipertrofia septal transitoria). La vigabatrina, por otro lado, puede causar efectos adversos como el estado encefalopático, pero la afectación retiniana no siempre se observa.
Intervención Neuropsicológica
La intervención neuropsicológica debe estar estrechamente ligada a los diagnósticos en curso, buscando mejorar la autonomía y calidad de vida. La literatura aún no recoge estudios que hayan aplicado un programa de intervención neuropsicológica específico para personas con síndrome de West y autismo asociado, por lo que el diseño de programas es fundamental.
Se propone un programa de intervención neuropsicológica dirigido a pacientes de entre 7 y 12 años con síndrome de West y TEA asociado, con una duración de seis meses. Este programa se estructuraría en dos sesiones individuales por semana de una hora, sumando un total de 48 sesiones (9 de evaluación y 39 de intervención). Se realizarían tres periodos de evaluación neuropsicológica: inicial, a mitad y al final de la intervención. Los dominios a evaluar son:
- Capacidad madurativa e intelectual general: Evaluada con la Escala de Inteligencia Wechsler (WISC-V) y el Cuestionario de Madurez Neuropsicológica para Escolares (CUMANES).
- Atención (sostenida y selectiva): Evaluada con el Test de Percepción de Diferencias Revisado (CARAS-R) y el Test de Colores y Palabras (Stroop).
- Funciones ejecutivas: Memoria de trabajo, inhibición, flexibilidad cognitiva, planificación y solución de problemas, evaluadas con el Test de Clasificación de Tarjetas de Wisconsin (WCST), el Test de Colores y Palabras (Stroop) y la Torre de Hanoi.
- Habilidades sociales: Evaluadas con la Escala para la Evaluación del Desarrollo Psicosocial (Hurtig y Zazzo).
Las 39 sesiones de intervención neuropsicológica incluirían actividades entremezcladas según criterios de variedad y complejidad, progresando desde la capacidad atencional hasta las funciones ejecutivas y, finalmente, las habilidades sociales y la solución de problemas. Se busca mantener un equilibrio entre la novedad y la práctica para mejorar la autoeficacia del paciente. Cada sesión de una hora contendría tres ejercicios diferentes de 15 a 20 minutos cada uno:
- Matriz de letras y números: Para la mejora de la atención sostenida y la planificación.
- Búsqueda del patrón: Para la mejora de la flexibilidad cognitiva, utilizando barajas de cartas.
- Sudokus: Para la mejora de la atención selectiva, memoria de trabajo y planificación.
- Solución de problemas: Tarea de lápiz y papel para mejorar la capacidad de solución de problemas y las habilidades sociales, abordando problemas sociales, familiares o económicos.
- Copia de una figura: Ejercicio de lápiz y papel para la mejora de la atención selectiva, memoria de trabajo y la impulsividad.
- Identificación de escenas: Para la mejora de las habilidades de interpretación de escenas sociales y comprensión emocional.
- Cómo me siento: Tarea dirigida a mejorar la identificación de las emociones y la gestión de la frustración.
- Lenguaje claro: Actividad dinámica para aumentar la capacidad de interacción social y el uso de un lenguaje conversacional adecuado.
Una en un Millón, Síndrome de West - Teleantioquia
Impacto en la Dinámica Familiar y Apoyo
El estrés emocional de ver a un bebé sufrir de espasmos y crisis epilépticas es extremadamente angustiante para los padres. Las demandas físicas y emocionales de cuidar a un niño con síndrome de West pueden alterar la rutina diaria y afectar las relaciones familiares. Además, el tratamiento a menudo incluye medicamentos, terapia y atención médica especializada, lo que puede generar costos elevados. Es fundamental que las familias reciban apoyo emocional y psicológico durante todo el proceso, tanto para afrontar las dificultades inmediatas como para hacer frente a los retos a largo plazo.