La progeria, o más específicamente el síndrome de progeria de Hutchinson-Gilford (HGPS), es un trastorno genético progresivo extremadamente raro y fatal, caracterizado por un envejecimiento brusco y prematuro que comienza tempranamente en la infancia. El término "progeria" proviene del griego pro ("hacia, a favor de") y geron o geras ("viejo"), significando "envejecer prematuramente". Esta enfermedad afecta a uno de cada cuatro millones de personas en el mundo y a uno de cada 20 millones de nacimientos, sin distinción de razas o género. Los niños con progeria suelen nacer con un aspecto saludable, pero desarrollan rápidamente características propias de la vejez, atrapados en cuerpos de ancianos.
Manifestaciones Clínicas y Síntomas Característicos
Los niños con progeria suelen parecer normales al nacer y no se diferencian del resto. Sin embargo, entre los 18 y 24 meses de edad o durante el primer año de vida, comienzan a manifestar una serie de rasgos distintivos y síntomas progresivos. Estos incluyen:
- Un retraso del crecimiento y falta de ganancia de peso, con una estatura baja.
- Pérdida del cabello, que conduce a una alopecia completa, así como la ausencia de cejas y pestañas.
- Alteraciones cutáneas, con un marcado envejecimiento prematuro de la piel que presenta arrugas, sequedad, esclerodermia, lesiones queratósicas y manchas hiperpigmentadas.
- Rasgos faciales muy característicos: ojos saltones o más grandes de lo habitual, venas muy marcadas en la superficie del cráneo, una cara afilada o "de pájaro", nariz prominente y afilada, y un mentón de menor tamaño (micrognatia). Las orejas suelen ser grandes.
- Rigidez, dislocación de la cadera y rango de movimiento limitado.
- Retraso o ausencia en la formación de los dientes.
- Una cabeza grande en proporción al tamaño de la cara (macrocefalia) y apertura de la parte blanda del cráneo (fontanela).
Es importante destacar que, a pesar de las severas manifestaciones físicas, los niños con progeria no presentan alteraciones neurológicas; son inteligentes, valientes y llenos de vida, y su desarrollo cognitivo y emocional no se correlaciona con el envejecimiento fenotípico. Otros órganos como el hígado, riñones, pulmón, sistema digestivo, médula ósea y cerebro no se ven afectados directamente por la enfermedad.

Bases Genéticas y Mecanismo Molecular
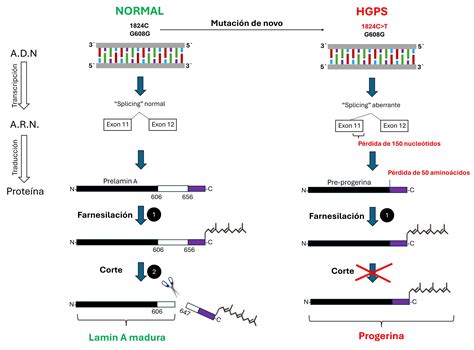
La progeria es una enfermedad genética debida a una mutación espontánea, lo que significa que los casos son esporádicos y generalmente no se heredan de los padres, sino que surgen de novo. Esta mutación puntual se localiza en el gen LMNA, situado en el cromosoma 1, que codifica para las proteínas lamina A y C. La lámina A es un componente fibrilar principal de la lámina nuclear, una estructura esencial que mantiene la forma del núcleo celular y participa en la organización de la cromatina, la replicación del ADN y la regulación del ciclo celular.
En el HGPS, la mutación más frecuente ocurre en la posición 1824 del exón 11 del gen LMNA, donde una citosina se sustituye por una timina. Esta mutación no cambia el aminoácido codificado, pero activa un punto críptico de corte en el procesamiento del ARN mensajero (ARNm). Como resultado, se produce una variante de prelámina-A más corta, con pérdida de 50 aminoácidos en el extremo carboxilo. Esta proteína anormal, conocida como progerina o lámina AD50, carece de un sitio de corte crucial para la enzima Zmpste24. Esto impide la correcta maduración de la prelámina-A en lámina A, lo que provoca que la progerina permanezca farnesilada constitutivamente.
La farnesilación es un proceso postraduccional normal en la prelámina-A, donde un grupo farnesilo (un lípido) se une a la cisteína terminal. Sin embargo, en la progerina, este grupo farnesilo no es removido. La acumulación de esta progerina farnesilada en el núcleo altera su estructura, provocando deformaciones, herniaciones y lóbulos. Estos defectos afectan la organización de la cromatina, predisponiendo a rupturas del ADN y a una señalización inadecuada para su reparación, lo que finalmente induce la senescencia o muerte celular acelerada.
Diagnóstico y Factores de Riesgo
La progeria suele aparecer en la infancia o en los primeros años de la niñez, con síntomas evidentes durante el primer año de vida. El diagnóstico se basa en un examen físico que identifica los rasgos característicos de la enfermedad. Las pruebas genéticas son fundamentales y pueden detectar los cambios en el gen LMNA. Se ha observado que, aunque es extremadamente rara, la edad del padre se ha descrito como un posible factor de riesgo, pero no se conocen factores ambientales o de estilo de vida que aumenten el riesgo de padecer progeria.
Un desafío significativo es el retraso en el diagnóstico, que puede tardar una media de cuatro años, y en un 20% de los casos, diez años o más. Este retraso impide la aplicación temprana de tratamientos preventivos o paliativos y dificulta el apoyo a las familias afectadas por la escasez de información y conocimiento sobre esta enfermedad. Actualmente, se tiene un registro de 203 menores en 50 países de todo el mundo que viven con progeria y laminopatías progeroides.
Complicaciones y Esperanza de Vida
El proceso de envejecimiento en pacientes con progeria se produce de 5 a 10 veces más rápido de lo habitual. La progeria se caracteriza por un endurecimiento grave de las arterias, conocido como aterosclerosis, que es una de las principales complicaciones. Las paredes de las arterias se endurecen y se hacen más gruesas, limitando el flujo sanguíneo.
La mayoría de los niños con progeria fallecen de enfermedades cardíacas, ataques al corazón o accidentes cerebrovasculares, que son las causas finales de muerte en la mayoría de los pacientes. La expectativa de vida media de un niño con progeria es de unos 13 años, oscilando en un rango entre los 7 y los 25 años. La gran mayoría no supera la adolescencia. Es notable que estos niños no suelen sufrir la enfermedad de Alzheimer, cataratas ni los cánceres típicos del envejecimiento.
Investigación y Avances Terapéuticos
Dada la naturaleza genética de la progeria debido a una mutación espontánea, no existen medidas preventivas conocidas. Sin embargo, la investigación ha avanzado significativamente en la última década.
La Fundación para la Investigación de la Progeria (PRF)
En 1998, los doctores Leslie Gordon y Scott Berns crearon la Fundación para la Investigación de la Progeria (PRF), motivados por el diagnóstico de HGPS de su hijo Sam. Esta organización, única en el mundo, promueve la investigación, el conocimiento de la enfermedad y recauda fondos. La PRF cuenta con un banco de células y tejidos de pacientes con progeria y sus familiares, que ha sido fundamental para el descubrimiento del gen causante en 2003 y para el desarrollo de nuevos estudios. Actualmente, la PRF tiene un registro de 49 líneas celulares de niños con progeria en todo el mundo.
Avances en Modelos Animales y Tratamientos Farmacológicos
El descubrimiento del gen causante del HGPS y el conocimiento de la patogenia molecular han propiciado investigaciones en modelos animales. Se han desarrollado ratones transgénicos con la misma mutación genética que los pacientes, que reproducen las manifestaciones clínicas de la progeria, lo que permite estudiar mecanismos moleculares y probar tratamientos.
Aunque actualmente no hay una cura, se han encontrado distintas posibilidades para bloquear el efecto nocivo de la progerina. Científicos de la Universidad de Durham (Reino Unido) descubrieron que la enfermedad es causada en parte por daños al ADN provocados por especies reactivas del oxígeno (ERO), y un fármaco llamado acetilcisteína-n parece controlar ese daño. En España, un grupo liderado por el Dr. Carlos López-Otín logró bloquear en ratones el desarrollo de la progerina, mejorando la condición cardíaca.
En 2020, la FDA aprobó el lonafarnib, un inhibidor oral de la farnesiltransferasa, como el primer medicamento disponible para tratar la progeria. Este fármaco ha demostrado prolongar la vida de los pacientes en un 30% de media. Las investigaciones actuales buscan evaluar nuevos tratamientos que, combinados con el lonafarnib, permitan incrementar tanto la esperanza como la calidad de vida de los niños.
Terapias del Futuro: Edición Genética CRISPR
El tratamiento definitivo consistirá en la eliminación de la mutación genética. Todavía falta tiempo para lograr esto de forma efectiva y segura en humanos, pero la tecnología CRISPR, una herramienta de edición genética, está siendo estudiada como un posible "corrector de errores" en el ADN. Científicos españoles han logrado revertir dicha mutación en un pequeño porcentaje de células en modelos murinos, logrando alargar hasta un 25% la vida del ratón transgénico progeroide. Estos avances son prometedores para futuras terapias génicas.
Descifrar el código secreto del envejecimiento | DW Documental
Casos Notables e Inspiradores
A lo largo de la historia, la progeria ha sido visibilizada a través de casos conmovedores y personalidades inspiradoras, que han impulsado la investigación y la concienciación.
Sam Berns: El Rostro de la Progeria
El caso de Sam Berns se convirtió en un símbolo global en la lucha contra la progeria. Sam, quien sufrió la enfermedad, fue el protagonista del documental de HBO "La vida según Sam" (2013). A pesar de su frágil aspecto, Sam mostró un impulso extraordinario y una voz inspiradora. "No quiero que cuando me vean la gente sienta lástima. He decidido dejar que me filmen para que la gente me conozca", expresó. Sam, que superó los 15 años (falleció a los 17), fue uno de los pocos pacientes que logró una longevidad excepcional para la enfermedad. Su historia destacó la importancia de vivir plenamente y la necesidad urgente de encontrar una cura, inspirando a científicos y al público.

Aaron Kushner: El Primer Caso Mediático
El nombre de la enfermedad se popularizó en 1981 con el libro "Cuando las cosas malas le suceden a la gente buena" del rabino Harold S. Kushner, escrito tras la muerte de su hijo, Aaron Kushner. Diagnosticado a los 3 años, Aaron a los 10 ya tenía el cuerpo de un hombre de 60. Murió dos días después de cumplir 14 años, con tan solo 11 kilos de peso, en 1977. Su padre prometió contar su historia para que no fuera olvidado, destacando el profundo impacto personal de la progeria.
Otros Casos de Resistencia y Visibilidad
- Rupesh Kumar, un niño indio, fue una de las personas con Hutchinson-Gilford más longevas del mundo, superando la barrera de los 20 años. Su familia rechazó ofertas inhumanas para que actuara en circos, demostrando la dignidad frente a la adversidad.
- En Colombia, Karen Ordóñez apareció en los noticieros hablando de sus problemas en la escuela, visibilizando las dificultades cotidianas de los niños con progeria. Karen decía que le encantaba tener amigos y que sus compañeras solían pensar que siempre estaba llorando cuando en realidad sus ojos lloraban constantemente por la falta de pestañas.
- El caso de Matías, a sus diez años, ilustra la lucha diaria. Con un cuerpo envejecido, él encuentra refugio en los libros y en la esperanza de la ciencia, enfrentando cada día con un coraje que muchos adultos envidiarían.
Estos testimonios subrayan la importancia de la financiación y la concienciación para acelerar el diagnóstico y el desarrollo de tratamientos. Aunque las investigaciones son lentas, cualquier avance en progeria es potencialmente extrapolable al envejecimiento normal de la población, generando un interés continuo en laboratorios e industria farmacéutica.