¿Qué es el Síndrome de Edwards?
El síndrome de Edwards, descrito en 1960, es un síndrome polimalformativo debido a la existencia de tres copias del cromosoma 18 en lugar de las dos copias normales. Nuestras células contienen 46 cromosomas, pero en las trisomías cada célula tendrá 47, pues hay tres copias de un cromosoma. Este cromosoma extra se expresa con retrasos serios en el desarrollo motor y cognitivo, y con múltiples malformaciones congénitas.
Tipos y Origen Genético
El 95% de los casos de síndrome de Edwards corresponden a trisomía completa, donde el cromosoma 18 adicional está presente en todas las células del individuo, ya sea por no disyunción o por translocación. La no disyunción es el evento que produce la trisomía 18 en la mayoría de los casos y ocurre en el espermatozoide o el óvulo que forma el feto, lo que significa que generalmente se produce de forma espontánea o por primera vez en ese embarazo, no siendo usualmente heredada. En casos muy poco frecuentes, el material extra puede adherirse a otro cromosoma (translocación).
También se describe un genotipo incompleto en el 5% de los casos, pudiendo ser:
- Trisomía 18 en mosaico: la presencia de un cromosoma 18 adicional en algunas células.
- Trisomía 18 parcial: la presencia de una parte de un cromosoma 18 adicional en las células.
Incidencia y Factores de Riesgo
El síndrome de Edwards es la segunda trisomía más frecuente después del síndrome de Down. Se presenta en aproximadamente 1 de cada 6.000 a 7.000 nacidos vivos, aunque esta estadística aumenta a 1 de cada 2.500 si se hace referencia al número de embarazos afectados, dependiendo de la realización de diagnóstico prenatal. Se produce en aproximadamente 4,1 de cada 10.000 embarazos (según los datos de abortos inducidos, muertes fetales y nacidos vivos). Aparece con mayor frecuencia en mujeres (una relación de 3:1 o 4:1) y está asociado a la edad avanzada de la madre, siendo el cromosoma extra casi siempre de origen materno.
Manifestaciones Clínicas y Anomalías Asociadas

El síndrome de Edwards presenta numerosas y variadas anomalías congénitas, lo que dificulta la generalización debido a que cada caso es distinto. Los bebés afectados suelen tener múltiples problemas y rasgos faciales particulares. La trisomía 18 se asocia con discapacidad intelectual, tamaño pequeño al nacer y diversas malformaciones congénitas.
Características Físicas y Dismórficas
- Retraso del crecimiento: Marcado retraso del crecimiento prenatal y posnatal, con bajo peso al nacer.
- Craneofaciales: Cabeza pequeña (microcefalia), occipital prominente, nuca prominente, mandíbula pequeña (micrognatia), orejas de implantación baja y malformadas, pabellones auriculares puntiagudos, hendiduras palpebrales cortas, boca pequeña, labio hendido, fisura en el paladar, hipertelorismo, cejas pobladas con sinofris, hipertricosis, hipoplasia de las fosas nasales. Los rebordes orbitarios son hipoplásicos, y estas características confieren un aspecto triangular a la cara. La plagiocefalia también se ha observado en algunos casos.
- Extremidades: Puños cerrados con el dedo índice sobre el dedo mayor y el meñique sobre el anular, o el segundo y quinto dedos superpuestos al tercero y cuarto. Uñas insuficientemente desarrolladas o hipoplásicas. Pies con un fondo redondeado (pies en mecedora o pies convexos), pies zambos, equinovaros o talo-valgo con atrofia muscular marcada. Piernas cruzadas. El dedo gordo del pie está acortado y a menudo en dorsiflexión. Puede haber hueso del radio ausente o malformado, contracturas articulares, retracciones en rodillas y tobillos y desviación ulnar de las manos. Se ha observado hipocratismo digital.
- Tórax: Esternón corto, forma inusual del pecho (tórax en quilla o cuadrado), pelvis estrecha.
- Piel: Pliegues cutáneos redundantes, especialmente en la nuca.
- Otros: Testículo no descendido en varones.
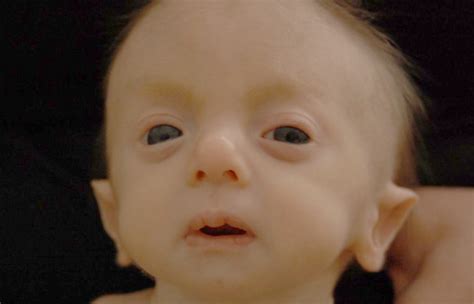
Malformaciones Orgánicas y Funcionales
- Cardiopatías congénitas: Son muy frecuentes (presentes en el 90% de los casos) y graves, incluyendo persistencia del conducto arterioso, comunicaciones interventriculares e interauriculares, coartación de aorta, ausencia del septo membranoso, y en casos severos, Tetralogía de Fallot.
- Malformaciones urogenitales y gastrointestinales: Riñón en forma de herradura, gran dilatación de la vía urinaria, ausencia de riñón. Interrupción del esófago (atresia de esófago), divertículo de Meckel, tejido pancreático ectópico, defectos del ano y recto, defectos de la pared abdominal (como onfalocele, donde los intestinos nacen fuera de la barriga), e intestinos en posición anómala.
- Sistema Nervioso Central (SNC): Retraso psicomotor y mental, discapacidad intelectual, alteraciones del tono muscular (hipotonía central severa), ventriculomegalia, falta de desarrollo del cerebelo y del cuerpo calloso (disgenesia del cuerpo calloso), y falta de formación de los dos hemisferios cerebrales. Aproximadamente el 30-50% de los pacientes con trisomía 18 tienen convulsiones (generalizadas, focales o mixtas).
- Problemas respiratorios: Hiperreactividad bronquial, rinitis obstructiva, bronquitis, neumonías de repetición, y apnea central.
- Musculares: Hernias o diastasis de los músculos rectos de la pared abdominal.
- Otras: Dificultades en la alimentación, estreñimiento persistente, sordera, problemas de la vista, escoliosis progresiva en niños mayores.
Diagnóstico del Síndrome de Edwards
El diagnóstico de la trisomía 18 se puede sospechar antes del nacimiento y confirmar mediante pruebas genéticas.
Diagnóstico Prenatal
El diagnóstico prenatal se realiza con pruebas citogenéticas. Se puede sospechar en la ecografía fetal mediante la detección de malformaciones anatómicas y otros marcadores:
- Hallazgos ecográficos: Polihidramnios, placenta pequeña, una sola arteria umbilical, restricción del crecimiento fetal, apariencia quística septada a nivel de la nuca, edema de pie generalizado, derrame pericárdico, probable defecto del tabique interventricular, angulación de pie respecto a las piernas, quiste del plexo coroideo, defectos del tubo neural, ventriculomegalia, cráneo en forma de fresa, cisterna magna anormal, cuerpo calloso ausente o hipoplasia cerebelosa. El aumento de la translucencia nucal (en el primer trimestre) o del pliegue (en el segundo) es el marcador ecográfico más confiable y ampliamente utilizado de las trisomías.
- Estudios Doppler: Regurgitación tricuspídea y flujo reverso en el ductus venoso.
- Cribado prenatal no invasivo: Análisis de ADN fetal libre de células en una muestra de sangre materna. Sin embargo, su sensibilidad y especificidad para la trisomía 18 es relativamente baja en comparación con la trisomía 21, por lo que las decisiones de manejo no deben basarse únicamente en sus resultados.
- Pruebas invasivas de confirmación: Se realizan mediante amniocentesis o biopsia de vellosidades coriónicas, con análisis citogenético del cariotipo, hibridación fluorescente in situ (FISH) y/o análisis de micromatrices cromosómicas. Es importante destacar que un hallazgo de trisomía 18 en la biopsia de vellosidades coriónicas puede requerir una investigación adicional, ya que la trisomía podría estar limitada a la placenta (mosaicismo placentario).
Diagnóstico Posnatal
Después del nacimiento, el diagnóstico se sospecha por la apariencia clínica del neonato y se confirma mediante pruebas en sangre periférica, realizando un cariotipo con bandeo G.
Pronóstico y Supervivencia
Luz y su Lucha con el Síndrome de Edwards: Conoce su Inspiradora Historia de Vida. CUN
El síndrome de Edwards se caracteriza por una alta tasa de mortalidad perinatal y en la primera infancia. La supervivencia media es inferior a un mes. Más del 50% de los niños fallecen durante la primera semana de vida, y menos del 10% sobrevive más allá del primer año, un dato que no se ha modificado significativamente con el tiempo. Las principales causas de defunción son la apnea central y las neumonías.
Sin embargo, la trisomía 18 no es completamente letal, y existen casos excepcionales de supervivencia prolongada, incluso hasta la adolescencia. En un estudio poblacional en Estados Unidos, la supervivencia a los cinco años fue del 12%, y aquellos que sobrevivieron al primer año de vida tuvieron una mayor probabilidad de alcanzar los cinco años.
Factores Asociados a Mayor Supervivencia
Aunque no se conocen con exactitud todos los factores que contribuyen a una supervivencia prolongada, se han asociado algunos elementos:
- Sexo femenino: Las niñas con trisomía 18 tienen mayor probabilidad de supervivencia por periodos más largos.
- Raza: Se ha asociado a raza distinta a la blanca.
- Mosaicismo: Aunque se ha postulado, no se ha demostrado una mayor supervivencia en los casos de mosaicismo.
- Cuidados y apoyo: Una intensa dedicación al niño, cuidados adecuados, y un fuerte apoyo psicológico y social por parte de la familia pueden influir positivamente en la longevidad y calidad de vida.
Complicaciones en Supervivientes a Largo Plazo
Los pacientes con una supervivencia prolongada, aunque infrecuente, suelen enfrentar problemas significativos:
- Alta dependencia psicomotora.
- Dificultades en la alimentación.
- Estreñimiento persistente.
- Infecciones de repetición.
- Escoliosis progresiva.
- Cardiopatía congénita grave.
- Riesgo aumentado de tumores de órganos sólidos (p. ej., hepatoblastoma, tumor de Wilms).
Manejo y Tratamiento
No existe un tratamiento específico que cure la anomalía genética subyacente de la trisomía 18. El manejo se centra en el tratamiento paliativo y sintomático, adaptado a las afecciones individuales de cada persona y a las preferencias familiares. A menudo se requiere un enfoque multidisciplinario.
Intervenciones Médicas y Rehabilitadoras
Las opciones de tratamiento recomendadas incluyen tanto cuidados paliativos como intervenciones que buscan prolongar la vida y mejorar la calidad de vida:
- Soporte para dificultades de alimentación: Puede requerir alimentación por sonda nasogástrica o gastrostomía.
- Manejo de problemas respiratorios: Fisioterapia respiratoria para facilitar el despegue y drenaje de secreciones, y soporte no invasivo si es necesario.
- Control de síntomas: Tratamiento del dolor, manejo de las crisis convulsivas.
- Rehabilitación: La derivación temprana a fisioterapia, logopedia y terapia de la alimentación es crucial. El tratamiento fisioterápico puede incluir estimulación global motora y sensorial, normalización del tono muscular, cinesiterapia suave poliarticular de miembros, flexibilización raquídea y estimulación del control cefálico.
- Medidas ortopédicas: Tratamiento postural para evitar la progresión de deformidades, medidas ortopédicas posturales, y en algunos casos, férulas o yesos.
- Prevención de complicaciones: Medidas de prevención y tratamiento precoz de las complicaciones. Se recomienda un control de cribado regular para detectar tumores de órganos sólidos, dado el mayor riesgo en estos pacientes.
Debate sobre Intervenciones Invasivas
Existe controversia sobre la realización de múltiples procedimientos invasivos, como la cirugía en cardiopatías graves, debido a la alta mortalidad en el primer año de vida y la discapacidad severa. Algunos estudios sugieren que la supervivencia puede ser similar al tratamiento conservador. Sin embargo, otros estudios han evidenciado que la cirugía cardíaca se asocia con una mejor supervivencia en pacientes con trisomía 18, aunque el tratamiento siempre debe ser individualizado y considerar el riesgo-beneficio, así como los aspectos éticos y el consentimiento informado de la familia.
Consideraciones Éticas y Apoyo Familiar
El diagnóstico y manejo del síndrome de Edwards representan una situación compleja y delicada tanto para los profesionales de la salud como para las familias. Los padres de niños diagnosticados con este síndrome necesitan disponer de tiempo y soporte psicológico para asimilar la información, ya que habitualmente la enfermedad es grave. Los profesionales acompañan a los padres, transmitiendo la información de la forma más empática, ajustada y profesional posible.
El Duelo y la Toma de Decisiones
Las familias pueden experimentar un duelo anticipado, considerando el mayor riesgo de abortos espontáneos, muertes intrauterinas y pérdidas fetales. La muerte de un hijo es un "duelo especial", un hecho para el cual no hay palabras específicas en el idioma. Este sufrimiento psicológico es un proceso íntimo y personal que, si no se habilita a nivel social, profesional y familiar, puede llevar a problemas de salud mental como depresión, ansiedad, estrés postraumático o abuso de tóxicos.
Desde un paradigma médico, en ocasiones los bebés no reciben la atención médica adecuada, reduciéndose a "medidas de confort" que el equipo médico decide, sin contemplar los deseos de los padres. Ejemplos de esto incluyen la negación de atención en neonatología a bebés con diagnóstico prenatal de trisomía 18 o la limitación a cuidados paliativos sin intentar otras intervenciones vitales.
Es fundamental empoderar a las familias, brindándoles información y alternativas, escuchándolos y posibilitando que expresen sus emociones y preguntas. Esto les permite participar plenamente en la toma de decisiones respecto a su hijo, desde el diagnóstico prenatal hasta cualquier etapa de la vida. De hecho, un estudio reveló que el 97% de los padres refiere que su hijo con trisomía 13 o 18 enriquece a la familia.
La Ley de Simón (Simon's Law)
En Estados Unidos, en estados como Kansas y Texas, existe la "Ley de Simón" (Simon's Law). Esta ley prohíbe a cualquier profesional de la salud denegar o restringir medidas para prolongar la vida, o autorizar una orden de no reanimar (ONR) para un menor sin la autorización de sus padres. Además, si un padre lo solicita, se debe proporcionar información sobre la política de futilidad del hospital. Esta ley surgió de la experiencia de los padres de Simon, un bebé con trisomía 18 que falleció a los pocos meses.
Casos Relevantes de Supervivencia Prolongada
A pesar del pronóstico generalmente desfavorable, existen casos documentados de pacientes con síndrome de Edwards que han logrado una supervivencia notable, destacando la importancia de un enfoque pluridisciplinar y la dedicación familiar.
Uno de estos casos es el de una paciente con trisomía 18 completa que sobrevivió más de 15 años. Nacida a término mediante cesárea por depresión neonatal, presentaba múltiples rasgos dismórficos. Su cariotipo demostró una trisomía 18 (47,XX, +18). A lo largo de su vida, presentó cardiomegalia, afectación perihiliar, costillas adelgazadas, subluxación coxofemoral bilateral, dilatación de los ventrículos laterales, ductus permeable, comunicación interventricular restrictiva, ausencia del septo membranoso y coartación de aorta. Durante su infancia, sufrió hiperreactividad bronquial, rinitis obstructiva, infecciones urinarias, bronquitis, neumonías de repetición, crisis hipertensivas y cuadros convulsivos. Se detectó divertículo de Meckel y tejido pancreático ectópico, requiriendo intervención por el primero. Además, sufrió fracturas. Su retraso ponderoestatural, psicomotor y mental ha sido marcado, necesitando la implantación de una gastrostomía. Actualmente, presenta pobre conexión al medio, tetraparesia espástica y múltiples deformidades ortopédicas secundarias, como escoliosis severa, miembros en flexión y pies equinovaro-aductos. Recibe tratamiento fisioterápico domiciliario que ha logrado estabilizar sus deformidades y reducir las infecciones respiratorias.
Otro caso documentado es el de una paciente de ocho años en Pereira, Colombia, hija de padres no consanguíneos. Producto del sexto embarazo (con dos abortos espontáneos previos), presentó hipertelorismo, plagiocefalia, micrognatia con pabellones auriculares de implantación baja, fisura palatina, arco palatino estrecho, cejas pobladas con sinofris, hipertricosis, hipoplasia de las fosas nasales, cuello corto, escoliosis, luxación congénita de cadera bilateral, pie equinovaro izquierdo y pie talo-valgo derecho con atrofia muscular marcada, manos con desviación ulnar con dedos superpuestos e hipocratismo digital, retracciones en rodillas y tobillos, hipotonía y cianosis central. Se le diagnosticó Tetralogía de Fallot, comunicación interventricular e interauricular, aorta cabalgada y estenosis valvular pulmonar severa, lo que la mantuvo con bajos niveles de saturación de oxígeno. A pesar de la severidad de su cardiopatía congénita, no fue candidata a cirugía cardiovascular debido a su pronóstico. Esta paciente no ha presentado convulsiones y es completamente dependiente para las actividades básicas de la vida diaria, recibiendo manejo interdisciplinario y apoyo familiar constante. Su supervivencia de ocho años es notable, dado que menos del 4% de los casos sobreviven después del primer año de vida, especialmente con cardiopatías severas no intervenidas.
Estos casos demuestran la complejidad del síndrome y la variabilidad en su presentación y evolución, enfatizando la necesidad de un abordaje integral y personalizado.
tags: #discapacidades #de #sindrome #de #edwards