El síndrome de Treacher Collins (STC), también conocido como disostosis mandibulofacial o síndrome de Treacher Collins-Franceschetti, es una afección genética rara y congénita caracterizada por deformidades y anomalías en la cara. Esta patología fue descrita por primera vez en 1889, y en 1900, el oftalmólogo Edward Treacher Collins presentó dos pacientes con los síntomas distintivos, publicando uno de los primeros informes de casos.
Se trata de una enfermedad que todavía es desconocida para muchos y forma parte de las denominadas enfermedades raras (ER) o poco frecuentes, lo que hace referencia a la baja prevalencia de casos en la población. La incidencia anual estimada al nacimiento es de 1/50.000, aunque en algunas estimaciones se menciona 1 de cada 10.000 personas. Afecta a la manera en que se desarrollan los huesos de la cara antes del nacimiento.
Causas Genéticas y Patrones de Herencia
El STC es causado por una mutación genética, un cambio en el ADN de una persona. Esta alteración se localiza principalmente en el cromosoma 5, donde el gen TCOF1 (5q32-q33.1) es el encargado del desarrollo de la cara y la cabeza. Además, puede estar asociado a mutaciones en los genes POLR1C (6p21.1) y POLR1D (13q12.2). Estos genes, en ausencia de mutaciones, producen una proteína fundamental para el correcto desarrollo de los huesos y tejidos blandos de la cara.
Las causas de esta anomalía pueden ser hereditarias, transmitidas por los progenitores, o por una mutación adquirida a lo largo del desarrollo embrionario. Estadísticamente, es más habitual el segundo caso, con un 60% de los fetos que sufren el síndrome por una mutación adquirida durante la embriogénesis, frente a un 40% de los casos que se deben a una mutación hereditaria.
En la mayoría de los casos, el STC es causado por una mutación "de novo", lo que significa que ninguno de los padres tenía el gen o sus síntomas. En estas mutaciones, el cambio en el ADN ocurre justo antes o después de la fecundación del óvulo por el espermatozoide.
- Cuando la mutación ocurre en los genes TCOF1 o POLR1D, la herencia sigue un patrón autosómico dominante. Esto implica que solo una copia del gen mutado es suficiente para que el individuo manifieste las características clínicas. En estos casos, los afectados tienen un 50% de riesgo de transmitir el síndrome a sus hijos.
- Si la mutación se encuentra en el gen POLR1C, la herencia es autosómica recesiva. Aquí, ambos padres son portadores de una copia mutada del gen sin manifestar la condición. Si ambos transmiten la copia mutada al niño, este desarrollará el síndrome. En estos casos, el riesgo de que los padres tengan un hijo afectado es del 25% en cada embarazo.
La gravedad de las manifestaciones clínicas puede variar considerablemente, incluso entre miembros de la misma familia afectados, lo que se conoce como diferencia en la penetrancia. Los síntomas pueden ser tan leves que un progenitor podría tener la mutación sin darse cuenta y transmitirla a un hijo con síntomas mucho más evidentes.
Manifestaciones Clínicas y Funcionales
El síndrome de Treacher Collins causa cambios que suelen ser simétricos, afectando principalmente al complejo orofacial, lo que compromete zonas como la cabeza, cara, boca y cuello, tanto en apariencia como en funcionalidad. Las características no siempre aparecen todas por igual; pueden manifestarse todas o solo algunas, y en mayor o menor medida, ya que cada persona presenta particularidades únicas.
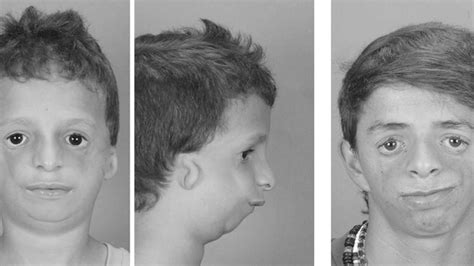
Deformidades Craneofaciales
Los rasgos distintivos incluyen:
- Hipoplasia malar (pómulos subdesarrollados).
- Colobomas de los párpados inferiores (defectos en el desarrollo de los párpados) y pestañas disminuidas, lo que puede causar problemas en la vista y exposición de las córneas al no poder cerrar los párpados completamente.
- Anomalías auriculares: Más del 50% de las personas con el síndrome nacen con microtia (orejas pequeñas o ausentes, con alteración en el oído externo). En el 77% de los casos se observan anomalías del oído externo, y en el 36% de estos, se detecta microtia o el estrechamiento de los canales auditivos externos. La definición latina de microtia es "oreja pequeña", refiriéndose a una deformidad congénita.
- Mandíbula muy pequeña (micrognatia) y poco desarrollada, así como un mentón retraído.
- Boca muy grande, paladar hendido o labio leporino.
- Orejas de implantación baja o anormalmente formadas, y conducto auditivo anormal.
- Vello del cuero cabelludo que se extiende hasta las mejillas.
Discapacidad Auditiva
La pérdida de audición es una de las características más frecuentes del síndrome. Es una discapacidad sensorial que se presenta desde el nacimiento, incluso con una ausencia casi total de dicho sentido en algunos casos. En el 40-50% de los casos, el síndrome es causa de hipoacusia transmisiva, que provoca pérdida o reducción auditiva debido a las alteraciones de la cadena de los huesecillos y a un desarrollo incompleto del oído medio. Las partes del oído interno tienden a preservarse en la mayoría de los casos.
Problemas Respiratorios y de Alimentación
Debido a la mandíbula pequeña y poco desarrollada (micrognatia), las vías respiratorias pueden estar menguadas, disminuyendo la capacidad respiratoria. Los recién nacidos pueden tener problemas para respirar porque sus vías aéreas son muy estrechas, lo que puede provocar apneas. Asimismo, algunos bebés presentan dificultades para alimentarse, especialmente cuando el síndrome interfiere con su respiración.

Implicaciones Funcionales y Motoras
Las alteraciones mencionadas previamente conllevan problemas que afectan directamente la función, lo que puede interpretarse como una discapacidad física y funcional que incluye aspectos motores.
- Dificultades en masticación y deglución: La anomalía de pómulos, mandíbula, mentón y dientes puede causar déficits de masticación y deglución. Para estos problemas, se interviene desde la fisioterapia y la rehabilitación.
- Retraso en el lenguaje y la comunicación: El niño puede presentar un retraso en el lenguaje y la capacidad de comunicación, así como dificultad para hablar, ya que el habla involucra una compleja coordinación motora orofacial.
- Anomalías en extremidades: En algunos casos, se requiere terapia para reconstruir o rehabilitar los pulgares, que pueden ser muy pequeños o inexistentes, lo que constituye una manifestación directa de discapacidad motriz en las extremidades.
Problemas Visuales
Las alteraciones de la condición conllevan también problemas que afectan a la vista, como úlceras en la córnea. Los niños con STC deben someterse a revisiones oculares regulares, ya que pueden tener problemas en la vista y en los movimientos oculares, debido a la exposición de sus córneas por la incapacidad de cerrar completamente los párpados.
Capacidad Intelectual
Es importante destacar que el síndrome de Treacher Collins no afecta la cognición; las personas afectadas tienen una inteligencia normal y no sufren ninguna modificación de sus cualidades intelectuales. Los niños con esta afección, en general, crecen hasta convertirse en adultos de inteligencia normal que se desempeñan bien.
Diagnóstico del Síndrome de Treacher Collins
El diagnóstico puede realizarse en diferentes etapas:
- Diagnóstico prenatal: En algunas ocasiones, es posible realizar un diagnóstico antes del nacimiento. Los genetistas pueden realizar un diagnóstico previo a la gestación para detectar posibles alteraciones cromosómicas en un futuro feto. La ecografía puede mostrar la curvatura de la mandíbula, lo que puede delatar la enfermedad. Las pruebas genéticas mediante amniocentesis y análisis de las vellosidades coriónicas también permiten un diagnóstico prenatal.
- Diagnóstico al nacer: El aspecto de la cara del niño al nacer suele hacer que los médicos sospechen el síndrome.
- Confirmación: Las radiografías de los huesos faciales del niño permiten identificar los rasgos característicos. Las pruebas genéticas confirman el diagnóstico al identificar las mutaciones en los genes TCOF1, POLR1C o POLR1D. Una vez diagnosticado, se puede profundizar en la evaluación con pruebas como TAC y rayos X para delinear el cuadro clínico.
Enfoque Terapéutico y Manejo Multidisciplinar
Aunque no existe una cura para el síndrome de Treacher Collins y la patología no se puede prevenir, una vez diagnosticada, es crucial intervenir con tratamientos quirúrgicos y otras herramientas para disminuir los síntomas y mejorar la calidad de vida. La atención y el cuidado de las personas con STC es continuo y requiere de la profesionalidad de un equipo multidisciplinario desde el nacimiento.
El tratamiento debe comenzar tan pronto como el niño nazca, especialmente si presenta problemas para respirar o alimentarse. La terapia sintomática se adapta a la gravedad de la enfermedad.
Síndrome de Treacher Collins
Intervenciones Quirúrgicas
Las personas con STC pueden precisar diversas intervenciones quirúrgicas a lo largo de su vida. Estas tienen como objetivo mejorar tanto la función como la apariencia facial, y se realizan durante una serie de años a medida que el niño crece. Incluyen:
- Cirugía maxilofacial para corregir deformaciones de paladar, pómulos, nariz, orejas, mandíbula y dientes.
- Reconstrucción de los párpados y avance de pómulos y mandíbula.
- Corrección de anomalías del oído.
- En casos de problemas respiratorios graves, puede ser necesaria una traqueotomía (introducción de un tubo en la tráquea) para mejorar la respiración.
- Se puede corregir el mentón retraído y otros cambios en la estructura de la cara mediante cirugía plástica.
Aunque las operaciones quirúrgicas evitan complicaciones futuras, la mayoría de las cirugías de reconstrucción facial se realizan cuando el niño es mayor, a menos que existan problemas respiratorios o de alimentación.
Terapias y Rehabilitación Funcional
Un equipo multidisciplinar que incluya neurocirujanos, otorrinolaringólogos, audiólogos, oftalmólogos, cirujanos plásticos y craneofaciales, cirujanos maxilofaciales, logopedas, educadores y psicólogos es fundamental.
- Rehabilitación auditiva: La audición se debe evaluar al nacer y regularmente. Puesto que el oído interno suele funcionar bien, los audífonos (que transmiten el sonido a través del hueso) pueden ser muy efectivos. Un logopeda puede informar sobre el uso y momento adecuados. La pérdida de audición se trata para garantizar un mejor desempeño escolar.
- Cuidado visual: Las alteraciones visuales se tratan con la ayuda de un oftalmólogo, ortoptista u óptico. Se recomiendan gafas especiales y adaptadas, incluso desde edad temprana, para evitar alteraciones como la ambliopía.
- Logopedia y comunicación: La logopedia (terapia del habla y el lenguaje) es a menudo necesaria, tanto si el niño presenta sordera como si no, para abordar el retraso en el lenguaje y la capacidad de comunicación.
- Fisioterapia para masticación y deglución: Para los problemas asociados a la masticación y deglución, se interviene desde la fisioterapia y la rehabilitación, buscando mejorar la función orofacial.
- Rehabilitación de pulgares: La terapia para reconstruir o rehabilitar los pulgares, que en algunos casos son muy pequeños o inexistentes, es una intervención clave para mejorar la función motriz de las manos.
- Odontopediatría: La actuación de un odontopediatra es importante para abordar problemas de infección, caries u ortodoncia, que requiere de un servicio muy especializado y coordinado con el equipo de cirugía maxilofacial.
- Estimulación temprana: Se resalta la importancia de la estimulación multisensorial durante los primeros años de vida.
Impacto Psicosocial y Calidad de Vida
Un diagnóstico con esta condición craneofacial tiene repercusiones significativas en la vida de la persona a nivel físico, social, educativo, psicológico y emocional. Por ello, la intervención temprana y un entorno comprensible y favorecedor son elementos clave.
Se ha demostrado que en niños y adolescentes con el síndrome, existen niveles elevados de ansiedad y angustia psicológica, lo que afecta su calidad de vida. La apariencia física o la estética repercute en el autoconcepto físico y el bienestar. Desde un punto de vista psicológico, los pacientes pueden enfrentar consecuencias como depresión y fobia social debido a las deformidades faciales, lo que disminuye su calidad de vida.
Es crucial fomentar entre la familia el cuidado y favorecer una planificación coordinada en la escuela, junto con estrategias de apoyo para las dificultades que presente la persona. Un estudio reveló que personas con una intervención adecuada y apoyo psicológico, además de un repertorio de habilidades de afrontamiento adquiridas, mostraron una calidad de vida intermedia y satisfactoria en dimensiones como salud física y psicológica, relaciones sociales y medio ambiente. Los participantes de otro estudio mostraron una lucha constante por la inclusión social y una gran motivación interna para aspirar a altas metas.
Concientización y Apoyo
El síndrome de Treacher Collins es una afección que dura toda la vida, y la persona que lo presenta debe hacer frente a muchas situaciones que ponen en riesgo su salud física y mental. Por ello, la comprensión hacia las experiencias psicosociales de quienes presentan esta condición debe ser la norma.
Existen recursos y figuras de superación que ayudan a comprender y visibilizar el STC. La película "Wonder" (2017) y el libro "Wonder. La Lección de August" de R. J. Palacio, han contribuido a la concienciación. Asimismo, se recomienda conocer documentales sobre la vida de Jono Lancaster. Organizaciones como FACES: The National Craniofacial Association, ofrecen información y apoyo.
Cada 28 de mayo se conmemora a nivel internacional el Día Mundial del Síndrome de Treacher Collins, una efeméride pensada para visibilizar la enfermedad, promover el entendimiento social y respaldar la investigación científica.