La discapacidad intelectual (DI) es una condición del neurodesarrollo que afecta del 1 al 3% de la población. Se caracteriza por un coeficiente intelectual (CI) cercano o menor a 70 y una edad de inicio antes de los 18 años.
Características de la Discapacidad Intelectual
Además del CI bajo y la edad de inicio temprana, la DI implica la incapacidad de adaptación funcional en al menos dos de las siguientes áreas:
- Comunicación
- Habilidades sociales / interpersonales
- Uso de recursos compartidos
- Autodirección
- Habilidades funcionales académicas, laborales, recreativas
- Salud y seguridad
La DI puede presentarse de forma aislada (no sindrómica) o junto con otras alteraciones médicas o del desarrollo (sindrómica). Esta distinción es crucial para un diagnóstico preciso y un manejo médico adecuado, que puede incluir el tratamiento de síntomas asociados.
El término "retraso global del desarrollo" (RGD) se utiliza para niños menores de 5 años que no cumplen los hitos esperados en dos o más áreas del desarrollo (social, motor o intelectual), con especial relevancia del lenguaje. Se estima que la prevalencia del RGD está entre el 1 y el 3 %.
Clasificación de la Discapacidad Intelectual
Cuando el CI es inferior a 70, se considera DI, y se divide generalmente en:
- Leve: CI entre 50 y 70
- Moderada: CI entre 35 y 50
- Grave: CI entre 20 y 35
- Profunda: CI inferior a 20
Causas Genéticas de la Discapacidad Intelectual
La etiología de la DI y el RGD es compleja, e incluye factores ambientales y genéticos, que pueden presentarse solos o en combinación. Las causas genéticas podrían estar implicadas en hasta el 50% de los casos. Las principales causas pueden resumirse en anomalías cromosómicas, anomalías del desarrollo del sistema nervioso central (SNC), teratógenos ambientales, DI sociocultural, complicaciones de la prematuridad, enfermedades monogénicas conocidas, causas sindrómicas y enfermedades metabólicas o endocrinas.
Existen muchas causas genéticas para esta condición, incluyendo mutaciones en genes que pueden heredarse bajo diferentes patrones: autosómico dominante, autosómico recesivo y ligado al cromosoma X. Dada la amplia variedad de causas genéticas, un abordaje con pruebas que abarquen un amplio número de genes puede ayudar a los médicos a realizar un diagnóstico preciso de forma más efectiva. Conocer la contribución genética no solo ayuda al tratamiento, sino que también permite refinar el consejo genético y definir los riesgos de recurrencia.
UNAM realiza diagnóstico genético de enfermedades raras
Identificación de Genes Relacionados con la Discapacidad Intelectual
Hasta el momento, los estudios genéticos han permitido identificar más de 1400 genes relacionados con la DI. La mayoría de estos genes codifican para proteínas, lo que hace que el análisis de exomas sea una aproximación habitual para el diagnóstico genético.
Un estudio genómico reciente, dirigido por investigadores de la Facultad de Medicina del Mount Sinai Hospital, ha identificado mutaciones en un gen no codificante (RNU4-2) relacionadas con la discapacidad intelectual. Este gen codifica para un ARN que forma parte de los espliceosomas, complejos encargados de procesar los ARNs mensajeros.
Los investigadores detectaron variantes poco frecuentes en el gen RNU4-2 en 47 personas, quienes compartían un patrón claro de síntomas:
- Discapacidad intelectual
- Microcefalia
- Corta estatura
- Hipotonía
- Convulsiones
- Retraso motor
Estos hallazgos resaltan la importancia de considerar la variación genética más allá de los genes que codifican para proteínas y permiten identificar un nuevo síndrome asociado a mutaciones en un gen concreto (que codifica para un ARN funcional), lo que puede ampliar el rendimiento diagnóstico de los pacientes con DI.
Nuevas Variantes Genéticas Identificadas
Estudios recientes han continuado identificando nuevas variantes genéticas asociadas a la discapacidad intelectual. Por ejemplo, en familias pakistaníes consanguíneas, se encontraron cuatro nuevas variantes en los genes FKRP, HIRA, BDH1 y TRAPPC6B, que cosegregaban con un patrón recesivo de herencia.
Además, variantes bialélicas de pérdida de función en la proteína transmembrana TMEM147 se han relacionado con discapacidad intelectual y espasticidad, como se observó en un estudio en Irán. Se cree que TMEM147 participa en la biogénesis de proteínas de membrana de paso múltiple.
En el contexto de trastornos del neurodesarrollo con epilepsia, se identificaron variantes homocigóticas en OCLN, ALDH7A1, IQSEC2 y COL3A1, y variantes previamente reportadas en CNTNAP2, TRIT1 y NARS1. La identificación de la variante en ALDH7A1, por ejemplo, ha demostrado utilidad clínica al dirigir el tratamiento con piridoxina.
Pruebas Genéticas Aplicadas al Diagnóstico de la Discapacidad Intelectual
La clave principal es lograr un diagnóstico preciso, probando una hipótesis clínica mediante la realización de las pruebas genéticas adecuadas. El análisis de ADN debe considerarse para cualquier persona con DI sin causa aparente.
Las pruebas genéticas se deben indicar cuando:
- Se haya realizado una exploración básica.
- Exista una historia familiar positiva de DI o RGD, de abortos o de mortinatos sin causa explicada.
- El retraso del desarrollo o mental se asocie con rasgos dismórficos, fenotipo conductual indicativo, regresión o consanguinidad.
- Se hayan descartado por completo otras causas.
El equipo de Genética Clínica de Clínica Alemana provee evaluaciones y asesoramiento a personas de toda edad y a sus familias, ofreciendo un análisis de 140 genes asociados a la discapacidad intelectual.
Listado de Genes Analizados en el Panel
El panel de genes asociado a discapacidad intelectual incluye los siguientes: ABCD1, ACSL4, ADNP, ALG13, ANKRD11, AP1S2, AP4B1, ARHGEF9, ARID1B, ARX, ATP7A, ATRX, BRWD3, CA8, CACNA1A, CASK, CC2D1A, CDKL5, CHD2, CHD7, CHD8, CNTNAP2, CREBBP, CTCF, CUL4B, DCX, DDX3X, DHCR7, DLG3, DNM1, DYNC1H1, DYRK1A, EHMT1, FGD1, FLNA, FMR1, FOLR1, FOXG1, FOXP1, FOXP2, FTSJ1, GAMT, GATM, GDI1,GNAO1, GPC3, GRIA3, GRIN1, GRIN2A, GRIN2B, HCN1, HDAC8, HNRNPU, HOXA1, HPRT1, HUWE1, IDS, IQSEC2, KAT6A, KCNJ10, KDM5C, KIAA2022, KIF1A, L1CAM, LAMP2, LINS, MAN1B1, MAOA, MBD5, MECP2, MED12, MED23, MEF2C, MID1, NDP, NDUFA1, NHS, NIPBL, NLGN3, NLGN4X, NRXN1, NSD1, NSUN2, OCRL, OFD1, OPHN1, OTC, PACS1, PAK3, PCDH19, PDHA1, PHF6, PHF8, PIGA, PIGN, PLP1, PNKP, POGZ, PORCN, PPT1, PQBP1, PTCHD1, PTEN, PTPN11, PURA, RAB39B, RAD21, RAI1, RPL10, RPS6KA3, SATB2, SCN2A, SCN8A, SLC16A2, SLC35A2, SLC6A1, SLC6A8, SLC9A6, SMARCA2, SMARCA4, SMARCB1, SMC1A, SMC3, SMS, ST3GAL3, STXBP1, SYN1, SYNGAP1, TBR1, TCF4, TIMM8A, TRAPPC9, TUSC3, UBE2A, UBE3A, UPF3B, VPS13B, WDR45, ZC4H2 y ZEB2. Estos genes también se incluyen en un panel más amplio de neurodesarrollo (Neurodevelopment-Expanded).
Beneficios de las Pruebas Genéticas
La confirmación molecular del diagnóstico puede ayudar a:
- Guiar las recomendaciones de tratamiento médico y tamizaje.
- Evitar exámenes y procedimientos innecesarios.
- Ofrecer consejo genético a la familia, incluyendo el diagnóstico prenatal en miembros asintomáticos en riesgo.
Evaluación Clínica y Asesoramiento Genético
El proceso de evaluación clínica es de gran importancia y debe seguir un protocolo ordenado. Un equipo médico especializado, compuesto por genetistas clínicos, neurólogos infantiles y asesores genéticos, realiza una evaluación inicial exhaustiva que revisa los antecedentes de la persona desde el embarazo y parto, el desarrollo, el crecimiento y los antecedentes de enfermedades.
Se aconseja traer toda la información médica disponible del paciente y sus familiares, como cuadernos de control del pediatra, resultados de exámenes previos (radiografías, ecografías, escáner, resonancia, estudios de laboratorio e informes de biopsias, entre otros). Los pacientes y familiares con antecedentes de una condición hereditaria se benefician del asesoramiento genético, obteniendo información y apoyo psico-emocional sobre la ocurrencia o riesgos de recurrencia de la enfermedad.
En la primera evaluación se practica un examen físico completo, detallado, sistemático y minucioso, y se toman fotografías del consultante y se evalúan imágenes de parientes para documentar los hallazgos. Basado en la información obtenida, el genetista considera las posibilidades diagnósticas y solicita exámenes adicionales si es necesario. Las conclusiones y recomendaciones se conversan en sesiones siguientes.
Tipos de Pruebas Genéticas Específicas
Cariotipo Convencional
Durante años, el cariotipo convencional fue la única herramienta para abordar las causas genéticas del retraso mental o del desarrollo. Permite un rastreo del genoma completo, pero está limitado a la detección de anomalías de un tamaño superior a 5 Mb. Su indicación debe aplicarse sin importar la presencia o ausencia de caracteres dismórficos, talla baja, anomalías congénitas o fenotipo conductual.
Test para Descartar el Síndrome del Cromosoma X Frágil
Este test consiste en el estudio molecular de la expansión CGC en el gen FRAXA. Debido a su bajo coste y alta prevalencia en pacientes con DI, se utiliza como prueba complementaria sistemática, aunque siempre se deben tener en cuenta algunos aspectos de la clínica.
Síndromes de Microdeleción
Para síndromes específicos, como el de Angelman o Prader-Willi, las técnicas de hibridación in situ fluorescente (FISH) son particularmente útiles. Utilizan sondas específicas de localización y su uso debe limitarse a casos donde el fenotipo sugiere un síndrome o enfermedad específica. En el síndrome de Prader-Willi, por ejemplo, si el FISH es normal, se indican estudios de metilación para descartar la unidisomía parental o mutaciones en el centro de imprinting.
Estudio de las Regiones Subteloméricas de los Cromosomas
El estudio de las regiones subteloméricas es de particular utilidad, estimándose que entre el 5 y 10% de los casos pueden diagnosticarse mediante estas técnicas, como la amplificación múltiple dependiente de ligamiento (MLPA) subtelomérica o FISH subtelomérico. Estas técnicas tienen una resolución mucho mayor que el estudio citogenético de alta resolución y son diez veces más sensibles.
El grado de retraso del desarrollo o mental es el principal factor predictor en este tipo de estudios. Retrasos moderados o graves asociados con dismorfia facial, anomalías físicas menores, baja talla y/o microcefalia están más probablemente asociados con anomalías subcromosómicas.
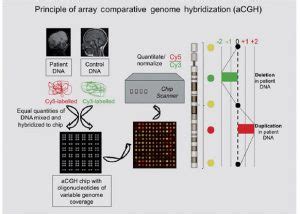
Microarrays de Hibridación Genómica Comparada (CGH-arrays)
Los CGH-arrays son un método alternativo para el rastreo de pequeñas pérdidas y/o ganancias de material genético, permitiendo la identificación de alteraciones inferiores a 1 Mb. Los estudios indican que con esta técnica se podrían diagnosticar un 10% más de casos de DI o RGD que con la combinación de estudios de citogenética convencional y/o FISH. Las recomendaciones actuales del American College of Medical Genetics (ACMG) sugieren su uso, junto con los tests citogenéticos habituales, para la evaluación de pacientes que presentan DI y anomalías congénitas.
Un estudio de 80 individuos con DI no explicada mostró que el array-CGH fue patológico en un 27,5% de los casos. Los factores asociados a un resultado patológico fueron antecedentes familiares de DI/RGD (OR: 12,1), malformaciones congénitas (OR: 5,33), más de 3 rasgos dismórficos faciales (OR: 20,9) e hipotonía periférica (OR: 3,25).
Las alteraciones cromosómicas más frecuentes detectadas fueron deleciones (62,5%), translocaciones no equilibradas (6,25%) y duplicaciones (31,25%). En el 51,6% de los análisis, las alteraciones cromosómicas se produjeron de novo.
Estudios de Genética Molecular
Los estudios de genética molecular permiten dirigir la investigación hacia una enfermedad muy concreta, como el estudio del síndrome del cromosoma X frágil (genes FraxA). La interpretación del tipo de herencia y la clínica del paciente pueden permitir establecer un juicio diagnóstico para indicar un test genético específico (por ejemplo, análisis molecular de LIS1 en el síndrome de Dandy-Walker o mutaciones RSK2 en el síndrome de Coffin-Lowry).
tags: #estudio #genetico #discapacidad #intelectual #y #anomalias