La progeria, también conocida como síndrome de progeria de Hutchinson-Gilford, es un trastorno genético progresivo extremadamente raro que provoca una aceleración del envejecimiento de los niños a partir de los primeros dos años de vida. Los niños afectados por la progeria suelen parecer sanos cuando nacen, pero pronto desarrollan una apariencia característica de envejecimiento acelerado, lo que a menudo lleva a que sean descritos como "bebés con cara de anciano".
Manifestaciones Clínicas y Síntomas Característicos
Generalmente, durante el primer año de vida, se comienza a notar que el crecimiento del niño es más lento. Los síntomas de este trastorno progresivo incluyen una serie de características físicas distintivas:
- Baja estatura.
- El cráneo de gran tamaño.
- Alopecia prematura (pérdida de cabello).
- Piel seca y arrugada.
- Ausencia de grasa subcutánea.
- Rigidez articular.
Además, estos niños pueden presentar características faciales propias como una cabeza más grande en proporción a su cara, mandíbula y boca pequeñas, labios finos, nariz en forma de pico y ojos grandes cuyos párpados no pueden cubrir completamente. A Sam, un niño diagnosticado con progeria, se le observó falta de crecimiento, pérdida de peso y cabello, arrugas y manchas en la piel, ojos saltones y rigidez. Su proceso de envejecimiento se acelera hasta siete veces más rápido que el de una persona sin esta condición. Es importante destacar que, mentalmente, los niños con progeria suelen seguir siendo acordes a su edad, ya que el cerebro no se ve afectado.
Causas Genéticas del Síndrome
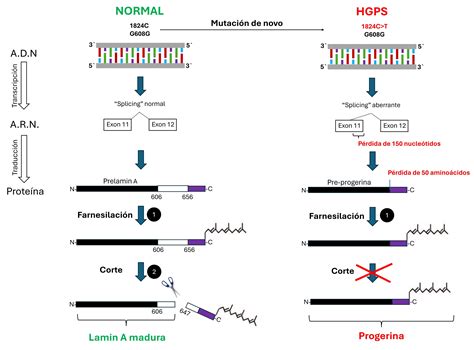
La progeria es causada por un cambio en un gen, conocido como lamina A. Este gen produce una proteína necesaria para mantener unido el centro de la célula, llamado núcleo. Cuando el gen lamina A experimenta un cambio, se produce una proteína lamina A defectuosa llamada progerina. Esta mutación, que es nueva y aparece por primera vez en una familia (denominada "de novo"), genera una proteína aberrante con 50 aminoácidos menos. Esta versión corta de la lámina A, la progerina, lleva además una molécula hidrofóbica por farnesilación, lo que la hace más tóxica y genera aberraciones en la estructura nuclear y en la expresión de genes desde el núcleo.
El gen alterado que causa la progeria rara vez se transmite de padres a hijos. Este descubrimiento constituye un punto de inflexión muy importante en la lucha contra esta enfermedad, permitiendo un diagnóstico más preciso y el estudio de los efectos de la progerina en tejidos y órganos.
Otros Síndromes Progeroides
Existen otros síndromes que pueden incluir problemas con proteínas similares a la progerina, conocidos como síndromes progeroides. A diferencia de la progeria de Hutchinson-Gilford, los genes alterados que causan estos síndromes sí pueden transmitirse de padres a hijos. Un ejemplo es el síndrome de Werner, también denominado progeria del adulto, que comienza en la adolescencia o al principio de la edad adulta.
Prevalencia y Expectativa de Vida
La progeria es una enfermedad extremadamente rara. Según el National Organization of Rare Disorders, la prevalencia es aproximadamente 1 en 20 millones, lo que significa que en un momento determinado hay aproximadamente 400 niños que viven con la enfermedad en todo el mundo. Sin embargo, hay medios especializados, como MedlinePlus, que difieren en el número y estiman que ocurre en 1 de cada 4 millones de recién nacidos. Hasta diciembre de 2020, el Registro Internacional de Progeria de la Progeria Research Foundation identificó un total de 131 niños y adultos jóvenes con progeria en el mundo.
Lamentablemente, las personas que padecen progeria no suelen pasar de los 13 años de edad. La mayoría de las personas que la padecen mueren cuando llegan a los 13-15 años. Viven, con suerte, poco más de 20 años, pasando su infancia atrapados en cuerpos de ancianos. Los problemas cardíacos o los accidentes cerebrovasculares son las causas finales de muerte en la mayoría de los niños que padecen progeria. La expectativa de vida media de un niño con progeria es de unos 15 años.
Complicaciones Asociadas
La progeria se caracteriza por un endurecimiento grave de las arterias, conocido como aterosclerosis. Esta es una enfermedad en la que las paredes de las arterias, vasos sanguíneos que transportan nutrientes y oxígeno desde el corazón al resto del cuerpo, se endurecen y se hacen más gruesas, lo que a menudo limita el flujo sanguíneo.
Casos Notables y Historias Personales
El nombre de la enfermedad se popularizó tristemente en 1981 con el éxito de ventas literario “Cuando las cosas malas le suceden a la gente buena” escrito por el rabino Harold S. Kushner tras la muerte de su hijo, Aaron Kushner, por progeria. A Aaron le diagnosticaron la enfermedad a los 3 años, y a los 10 su cuerpo ya era como el de un hombre de 60. Dos días después de cumplir 14, y con tan solo 11 kilos de peso, Aaron murió en 1977. Su padre prometió contar su historia para que no fuera olvidado.
Un caso de longevidad excepcional fue el de Rupesh Kumar, un niño indio del norte de la India, quien fue una de las personas con Hutchinson-Gilford más longevas del mundo. Con 20 kilos de peso, Rupesh superó la barrera de los 20 años. Su padre relató que los primeros síntomas de progeria en su hijo se dieron cuando Rupesh tenía dos años, comenzando con fuertes dolores de cabeza y estómago. Con el tiempo, la cabeza de Rupesh creció “anormalmente” y el niño empezó a perder peso de manera drástica, además de la constante caída de cabello y piel. La familia de Rupesh incluso recibió una inhumana oferta para que su hijo actuara en circos, la cual rechazaron categóricamente. Siete años antes, se creía que Rupesh era el superviviente de progeria de mayor edad tras la muerte del sudafricano Leon Botha a la edad de 26 años.
Tiene el tamaño de un niño y el rostro arrugado: el drama de un joven que envejece rápidamente
Otros casos incluyen a Bayezid Hossain, un niño con la misma enfermedad que nació hace unos años en un distrito muy cercano a uno de los casos recientes. En Latinoamérica, en 2016, se hizo famoso el caso de una niña que murió a los 12 años. En Colombia, Karen Ordóñez apareció en los noticieros hablando de sus problemas en la escuela a causa de su enfermedad. Ordóñez decía que le encantaba tener amigos y que sus compañeras solían pensar que siempre estaba llorando, cuando en realidad era que, debido a su falta de pestañas, sus ojos le lloraban constantemente.
Detecciones Recientes y la Reacción Familiar
Recientemente, un bebé con cara de anciano y abundante pelo en la espalda ha nacido en Bangladesh. Los padres del pequeño, Biswajit Patro y su esposa Parul, están encantados con el nacimiento de su hijo, al que califican de “milagro”. Aseguran que procurarán por todos los medios que crezca sano y lo van a aceptar tal y como es, sin sentir tristeza ni pena porque su hijo sea diferente.
En Israel, el primer caso de progeria se dio el año pasado en el Centro Médico Rambam de Haifa. Se trataba de una bebé de dos meses que había empezado a denotar “cosas inusuales en su piel” y presentaba características faciales propias de la progeria, como una cabeza más grande que su cara, mandíbula y boca pequeñas, labios finos, nariz en forma de pico y ojos grandes cuyos párpados no pueden cubrir.
Tratamientos y Futuras Promesas
Actualmente, para la progeria aún no hay una cura definitiva. Sin embargo, se han explorado y aplicado tratamientos experimentales. El proceso de envejecimiento del bebé de Israel fue tratado con lonafarnib, un inhibidor oral de la farnesiltransferasa, el único fármaco disponible hasta el momento para tratar la progeria y que recién puede ser utilizado en pacientes de al menos un año.
El Dr. Yugantar Pandey, profesional que atendió a Rupesh, indicó que la mayoría de las personas que padecen progeria mueren a los 13-15 años, pero en algunos casos, como el de su paciente, pueden vivir hasta los 21. La Dra. Hiba Zaaroura, médica israelí que atendió un caso reciente, subrayó la necesidad de un seguimiento cercano por un cardiólogo debido al “riesgo de sufrir problemas cardíacos y accidentes cerebrovasculares” y por un gastroenterólogo por “problemas de crecimiento”.
Gracias a la investigación iniciada por fundaciones como la creada por los padres de Sam en 1999, que permitió descubrir la causa molecular de la enfermedad en 2003 (la proteína progerina), los científicos pueden estudiar sus efectos. La creación de un ratón transgénico con la misma mutación genética que Sam ha sido clave, ya que estos ratones desarrollan las mismas manifestaciones clínicas. Esto ha permitido el desarrollo experimental de varios tratamientos, incluyendo terapias que evitan la farnesilación, es decir, la inclusión de la molécula hidrofóbica que lleva la lámina A aberrante.
El tratamiento definitivo consistirá en la eliminación de la mutación genética. Aunque todavía falta tiempo para lograrlo de forma efectiva y segura en humanos, científicos españoles han dado un paso importante. Utilizando la técnica de edición genética CRISPR, han logrado revertir dicha mutación en un pequeño porcentaje de células, alargando hasta un 25% la vida del ratón transgénico progeroide.
La Progeria y "El Curioso Caso de Benjamin Button"
A menudo, los niños con progeria son comparados con "Benjamin Button" de la vida real. “El curioso caso de Benjamin Button”, el cuento de Francis Scott Fitzgerald de 1922 (y su adaptación cinematográfica de 2008 con Brad Pitt), narra la historia de un hombre que nace viejo y poco a poco va rejuveneciendo. Tristemente, ninguno de los casos reales de progeria experimenta este proceso inverso de rejuvenecimiento con el correr del tiempo.