Las epilepsias mioclónicas progresivas (EMP) representan un conjunto de enfermedades neurodegenerativas raras que se cuentan entre las formas más discapacitantes de epilepsias. Aunque poseen una marcada heterogeneidad clínica y genética, comparten características comunes como la presencia de mioclonías de acción, crisis epilépticas y un deterioro neurológico progresivo. Estas condiciones suelen manifestarse en la infancia tardía o la adolescencia, pero pueden afectar a individuos de cualquier edad.
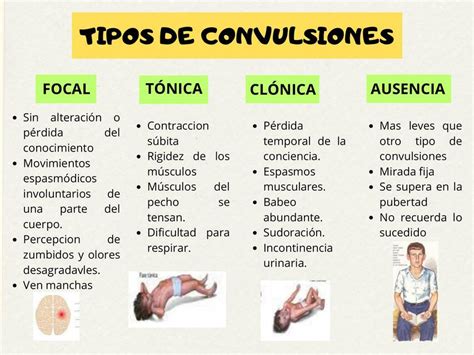
Las mioclonías, que dan nombre a estas enfermedades, son contracciones musculares súbitas y muy breves, resultado de descargas anómalas en el sistema nervioso central, cuyo origen es usualmente cortical o cortico-subcortical. Se distinguen por ser desencadenadas por la actividad muscular voluntaria, ciertos estímulos sensitivos o emociones. En cuanto a las crisis epilépticas, predominan las tónico-clónicas generalizadas, aunque pueden presentarse otros tipos como ausencias, crisis tónicas o focales. Es común observar un enlentecimiento de fondo en el electroencefalograma (EEG).
Actualmente, la etiología genética de la mayoría de las EMP es conocida, con una herencia típicamente autosómica recesiva, si bien existen formas autosómicas dominantes (como la atrofia dentato-rubro-pálido-luisiana) o mitocondriales (como la epilepsia mioclónica con fibras rojas rasgadas o MERRF). La identificación genética del síndrome es crucial, ya que algunos pacientes pueden beneficiarse de tratamientos específicos, como en el caso de la enfermedad de Gaucher o de Tay-Sachs. No obstante, aún existen pacientes en los que no se logran identificar variantes genéticas patogénicas, impidiendo su clasificación. El pronóstico de estos pacientes es variable según la enfermedad específica, pero a menudo es desfavorable, conduciendo a la discapacidad o a una muerte temprana.
Tipos de Epilepsias Mioclónicas Progresivas (EMP) y su Impacto en la Discapacidad
A continuación, se revisan algunas de las formas más frecuentes de epilepsia mioclónica progresiva y su sintomatología, que a menudo conlleva diversos grados de invalidez.
Epilepsia Mioclónica de Unverricht-Lundborg (EMP1)
La EMP1 es la epilepsia mioclónica progresiva más prevalente a nivel global. En Finlandia, por ejemplo, se describe una prevalencia de uno en 20,000 nacimientos. Su inicio suele ser con crisis tónico-clónicas generalizadas (CTCG), especialmente al despertar o durante el sueño, acompañadas de mioclonías que también predominan al despertar. Con la progresión de la enfermedad, las mioclonías se intensifican, se asocian al movimiento y aumentan con el estrés. Característicamente, pueden ser desencadenadas por estímulos sensoriales como luces o el tacto, o por la sorpresa, y pueden evolucionar a convulsiones en cascada, culminando en una CTCG breve. Estas manifestaciones pueden llevar a una severa discapacidad, llegando a postrar al paciente en una silla de ruedas.
- Genética: Enfermedad hereditaria autosómica recesiva, causada por variantes en el gen CSTB, que resulta en una deficiencia de cistatina B.
- Clínica: Además de las mioclonías y CTCG, los pacientes desarrollan síntomas cerebelosos como ataxia, incoordinación, temblor de intención y disartria. Las alteraciones cognitivas son generalmente ausentes o leves a moderadas.
- Diagnóstico: Se basa en el estudio genético para identificar variantes patogénicas en el gen EMP1, en conjunto con la sospecha clínica y los hallazgos del EEG.
- Manejo: El valproato es el tratamiento antiepiléptico más común. El piracetam puede aliviar las mioclonías. Se desaconseja el uso de lamotrigina y fenitoína por sus posibles efectos adversos. Otros fármacos incluyen fenobarbital, primidona, levetiracetam, topiramato y zonisamida. Las benzodiacepinas pueden usarse con precaución. En casos intratables, se consideran la estimulación del nervio vago o cerebral profunda, aunque con evidencia limitada.
- Pronóstico: La enfermedad progresa durante 5-10 años y luego se estabiliza. El impacto en la vida del paciente varía desde una vida independiente hasta la dependencia total en silla de ruedas o la postración en cama.
Enfermedad de Cuerpos de Lafora (EMP2)
También conocida como epilepsia mioclónica progresiva tipo II, la enfermedad de Lafora es una de las formas más devastadoras de EMP, llevando a un rápido deterioro y una profunda invalidez.
- Genética: Hereditaria autosómica recesiva, causada por variantes en los genes EPM2A (codifica la laforina) o EPM2B (codifica la malina). Se ha propuesto un tercer gen, PRMD8, asociado a una forma de inicio temprano.
- Epidemiología: Mayor prevalencia en pacientes con ascendencia mediterránea, del norte de África o Asia central, con aproximadamente 4 casos por millón de personas.
- Clínica: Los síntomas iniciales son crisis epilépticas occipitales focales o generalizadas, a menudo desencadenadas por la luz. De forma progresiva, las CTCG aumentan en frecuencia y la mioclonía se vuelve casi continua, especialmente en las extremidades, postrando al paciente en silla de ruedas. El deterioro neurológico y cognitivo es progresivo y rápido, manifestándose como disminución del rendimiento académico, afasia, apraxia, ataxia cerebelosa, hipotonía, espasticidad y ceguera cortical.
- Diagnóstico: Se basa en hallazgos clínicos, EEG e histológicos (presencia de cuerpos de Lafora en biopsias de glándulas sudoríparas). El análisis genético confirma variantes en EPM2A o EPM2B.
- Manejo: Fármacos como valproato, fenobarbital, benzodiacepinas, piracetam, levetiracetam y zonisamida son comunes. Se deben evitar vigabatrina, carbamazepina, fenitoína, gabapentina, pregabalina, tiagabina y lamotrigina. El perampanel ha mostrado beneficios.
- Pronóstico: Los pacientes terminan postrados en cama debido a la severidad de la progresión.
Síndrome de Falla Renal con Mioclonías (EMP4)
Conocida también como epilepsia mioclónica progresiva tipo 4, esta condición destaca por la combinación de síntomas neurológicos y renales que, en conjunto, pueden generar una importante incapacidad.
- Genética: Enfermedad autosómica recesiva ligada a variantes en el gen SCARB2, que codifica la proteína LIMP2.
- Epidemiología: Típicamente, el inicio ocurre entre los 15 y 25 años, aunque existe una forma de inicio tardío sin falla renal.
- Clínica: Gran heterogeneidad genotipo-fenotipo. Los pacientes pueden presentar inicialmente sintomatología renal (proteinuria, daño renal progresivo) o neurológica (temblores en dedos y manos que progresan a mioclonías de acción, mioclonías involuntarias en reposo y finalmente CTCG). Las mioclonías son multifocales y afectan tronco, extremidades y cara. Otros síntomas incluyen ataxia, disartria, alteraciones auditivas y neuropatía periférica desmielinizante.
- Diagnóstico: Se sospecha en pacientes con epilepsia mioclónica que desarrollan falla renal. El estudio genético identifica variantes en el gen SCARB2. El EEG muestra anormalidades epileptiformes generalizadas.
- Manejo: Se utiliza un régimen múltiple con valproato, clonazepam, barbitúricos y piracetam, con efectividad parcial. Debe evitarse lamotrigina, carbamazepina y oxcarbazepina.
Epilepsia Mioclónica con Ataxia y Función Cognitiva Normal (PRICKLE-1)
- Genética: Causada por variantes en el gen de la proteína de polaridad de la célula planar 1 (PRICKLE-1).
- Epidemiología: La edad de inicio es entre 5 y 10 años.
- Clínica: Se caracteriza por crisis epilépticas mioclónicas, CTCG (a menudo relacionadas con el sueño), ataxia y una función cognitiva normal. La ataxia precede a las crisis mioclónicas. Las mioclonías de acción afectan los músculos de las extremidades o bulbares, a veces con mioclonías espontáneas faciales que causan disartria. Pueden aparecer signos de neuropatía periférica.
- Diagnóstico: En niños o adolescentes con esta sintomatología, se deben descartar primero la enfermedad de Unverricht-Lundborg y los cuerpos de Lafora.
- Manejo: El tratamiento antiepiléptico puede incluir valproato, clonazepam, zonisamida, piracetam y levetiracetam. Se recomienda evitar fenitoína, carbamazepina y oxcarbazepina.
Epilepsia Mioclónica Progresiva del Mar del Norte (GOSR2)
- Genética: Causada por una variante en el gen del complejo receptor de Golgi SNAP 2 (GOSR2).
- Epidemiología: Se inicia alrededor de los 2 años. Hasta el momento, solo se han descrito 20 casos.
- Clínica: La ataxia y la arreflexia se inician alrededor de los 2 años, seguidas de mioclonías a los 6-7 años y crisis epilépticas diversas a los 13-14 años. En la segunda década de vida, los pacientes pierden la independencia ambulatoria debido a las crisis epilépticas, lo que genera una marcada incapacidad motora. Las mioclonías son generalizadas, fotosensibles y empeoran con la actividad o el estrés. En etapas tardías, hay deterioro cognitivo. La mayoría desarrolla escoliosis en la adolescencia y otras deformidades óseas.
- Manejo: El tratamiento con valproato, zonisamida, piracetam y levetiracetam suele no ser efectivo.
Epilepsia Mioclónica con Fibras Rojas Rasgadas (MERRF)
Conocido por sus siglas en inglés, MERRF, esta enfermedad mitocondrial también conduce a un grave deterioro y discapacidad.
- Genética: Causada por mutaciones en el ADN mitocondrial, principalmente en el gen MT-TK (tRNALys3), responsable del 80% de los casos.
- Clínica: El inicio suele ser en la niñez, con un desarrollo normal hasta la aparición de las mioclonías. Otras manifestaciones comunes incluyen epilepsia generalizada, ataxia, debilidad muscular, demencia, estatura baja, pérdida de audición y atrofia ocular. Síntomas más raros son cardiomiopatía, retinopatía pigmentaria y lipomas múltiples.
- Diagnóstico: La sospecha se basa en cuatro síntomas canónicos: mioclonías, epilepsia generalizada, atrofia cerebelosa y fibras rojas rasgadas en una biopsia muscular (coloración tricrómica de Gomori). Se confirma con estudio genético de genes mitocondriales. El electroencefalograma muestra... (información incompleta en el borrador original).
Epilepsia: ¿Cómo afecta en la vida cotidiana de quien padece esta enfermedad?
Epilepsia Mioclónica Juvenil (EMJ) y su Impacto a Largo Plazo
La epilepsia mioclónica juvenil (EMJ) es un síndrome epiléptico generalizado caracterizado por un inicio en la pubertad y tres tipos de crisis: mioclónicas, tónico-clónicas generalizadas (CTCG) y ocasionales ausencias típicas. Clásicamente, se ha considerado que requiere tratamiento antiepiléptico de por vida debido a la alta tasa de recaídas tras la suspensión de fármacos. Sin embargo, estudios recientes a largo plazo han cuestionado esta postura, sugiriendo la posibilidad de suspender el tratamiento exitosamente en ciertos pacientes.
Estudios descriptivos observacionales retrospectivos, incluyendo uno con pacientes de EMJ con más de veinte años de evolución, han investigado las características clínicas y la evolución. Se ha encontrado que un porcentaje de pacientes puede mantenerse libre de crisis sin tratamiento antiepiléptico durante periodos prolongados. La EMJ es reconocida como un trastorno heterogéneo con un amplio espectro fenotípico, lo que implica que el pronóstico y la respuesta al tratamiento pueden variar significativamente entre individuos.
Factores asociados con un mal pronóstico de libertad de crisis incluyen la presencia de los tres tipos de crisis, crisis mioclónicas precediendo a las tónico-clónicas generalizadas, comorbilidad psiquiátrica, y ciertas características del EEG. Por otro lado, un buen pronóstico se ha relacionado con mioclonías aisladas al inicio del cuadro, un tiempo prolongado hasta la primera CTCG y una duración corta de la epilepsia activa hasta la consecución de la libertad de crisis. La evidencia sugiere que mantener el tratamiento antiepiléptico con libertad de crisis de forma prolongada, con una retirada tardía, podría aumentar las posibilidades de una suspensión exitosa sin recaída.
La Epilepsia como Discapacidad: Aspectos Legales y Prestaciones
Si la epilepsia, especialmente sus formas mioclónicas progresivas, interfiere significativamente con la capacidad de una persona para trabajar o realizar actividades diarias, se considera una discapacidad. Organismos como la Administración del Seguro Social (SSA) en EE. UU. y la legislación española reconocen la epilepsia como una condición que puede otorgar derecho a beneficios, siempre que se cumplan ciertos criterios médicos y funcionales.
Síntomas y Desencadenantes que Afectan la Capacidad Funcional
Los síntomas de las crisis epilépticas pueden variar drásticamente y afectar la capacidad para mantener un empleo, conducir o completar tareas cotidianas, lo cual es central al determinar la discapacidad. Estos pueden incluir sacudidas repentinas e involuntarias, confusión, miradas fijas, pérdida de conciencia, fatiga post-convulsiva, pérdida de control muscular, alteraciones sensitivas y dificultades del habla.
Reconocer los desencadenantes es vital para el manejo y la documentación del impacto de la enfermedad. Algunos de los desencadenantes comunes son:
- Falta de sueño o fatiga
- Luces o patrones parpadeantes
- Estrés o tensión emocional
- Consumo de alcohol o drogas
- Fluctuaciones hormonales
- Omisión de medicamentos
- Enfermedad o fiebre
- Deficiencias nutricionales
- Ciertos medicamentos (p. ej., difenhidramina)
Criterios para la Discapacidad por Epilepsia
La SSA en Estados Unidos incluye la epilepsia como una condición que califica bajo su Libro Azul (Listado 11.02, Trastornos Neurológicos). Para recibir beneficios, se debe demostrar que la condición impide trabajar, incluso con tratamiento constante. Los requisitos específicos son:
- Convulsiones tónico-clónicas generalizadas: Al menos una vez al mes durante más de 3 meses consecutivos, a pesar del cumplimiento del tratamiento.
- Convulsiones discognitivas: Al menos una vez a la semana durante más de 3 meses consecutivos, a pesar del tratamiento.
- Convulsiones menos frecuentes con limitaciones funcionales significativas: Como funcionamiento físico limitado, dificultad de memoria, concentración o comunicación, problemas para interactuar con otros o adaptarse a situaciones nuevas.

Evaluación de la Capacidad Funcional Residual (CFR)
Si la epilepsia no cumple directamente con los criterios del listado, aún se puede tener derecho a una prestación por la evaluación de la Capacidad Funcional Residual (CFR). La SSA evalúa lo que la persona puede y no puede hacer física y mentalmente en un entorno laboral, considerando factores como edad, nivel de educación, experiencia laboral previa, otras condiciones médicas y limitaciones funcionales específicas (p. ej., no trabajar cerca de maquinaria o conducir). Para calificar, se debe demostrar que la epilepsia impide realizar cualquier tipo de trabajo a tiempo completo.
Epilepsia y el Empleo: Adaptaciones y Discriminación
Según la Ley de Estadounidenses con Discapacidades (ADA), la epilepsia es una discapacidad si limita sustancialmente actividades importantes de la vida. Los empleadores están obligados a proporcionar adaptaciones razonables, a menos que cause dificultades excesivas. Estas adaptaciones pueden incluir:
- Horarios flexibles para descanso o citas médicas.
- Evitar luces estroboscópicas o parpadeantes.
- Permiso para trabajar en entornos más tranquilos.
- Tiempo libre después de las convulsiones.
La discriminación por epilepsia es denunciable, y es importante hacerlo para prevenir futuros casos.
Tipos de Incapacidad Laboral y Grado de Discapacidad en España
En España, el tipo de incapacidad laboral concedida depende de cómo las secuelas de la epilepsia afectan la capacidad para trabajar. Las sentencias favorables a menudo resultan en:
- Incapacidad Permanente Total: Para la profesión habitual.
- Incapacidad Permanente Absoluta: Para cualquier profesión.
- Gran Invalidez: Cuando la epilepsia se acompaña de otras patologías que agravan el estado de salud y requieren la asistencia de otra persona para los actos esenciales de la vida.
Si existe un agravamiento de la epilepsia o una nueva patología, se puede solicitar una revisión de grado para aumentar la pensión. El importe de la pensión varía según el grado, calculándose sobre las bases de cotización. En el caso de Gran Invalidez, se añade un complemento significativo a la pensión.
El grado de discapacidad mide cómo la enfermedad afecta todos los aspectos de la vida diaria. Con una afectación moderada, se puede obtener un mínimo del 33%, y en casos graves, superar el 65%.
La dependencia evalúa la afectación de las secuelas en los actos básicos de la vida diaria (higiene, alimentación, desplazamiento). En casos avanzados de epilepsia que afectan gravemente múltiples órganos o funciones, se puede obtener cualquiera de los tres grados de dependencia: moderada, severa o gran dependencia.
Epilepsia: ¿Cómo afecta en la vida cotidiana de quien padece esta enfermedad?
Aspectos Prácticos y Legales de la Solicitud de Incapacidad
- Información a la empresa: No es obligatorio informar a la empresa sobre el trámite de incapacidad laboral, ya que es información confidencial.
- Solicitud durante la baja médica: No es necesario agotar los 18 meses de baja. Es incluso preferible presentar la solicitud antes para tener un mayor control del expediente.
- Jubilación por coeficientes reductores: Si no se han cumplido los 65 años, se puede solicitar la incapacidad laboral, lo que generalmente resulta en una pensión superior. Incluso tras los 65 años, si el hecho causante es anterior a la edad legal de jubilación, es posible recurrir a los tribunales.
- Compatibilidad con el trabajo: Es posible compatibilizar una pensión por incapacidad laboral con un trabajo, siempre que este esté adaptado a las secuelas de la enfermedad. Para una incapacidad total, no se pueden realizar las mismas tareas del trabajo anterior. Para la absoluta o gran invalidez, solo es viable en centros especiales de empleo y cumpliendo los trámites administrativos adecuados.
Evidencia Médica Esencial para Probar la Discapacidad
Para fortalecer el caso y demostrar que la epilepsia es una discapacidad, es crucial recopilar y presentar:
- Un diagnóstico formal de un neurólogo.
- Resultados de pruebas como EEG, resonancias magnéticas o tomografías computarizadas.
- Registros detallados de convulsiones (fechas, horas, síntomas).
- Notas del médico que describan la frecuencia y el impacto de las convulsiones.
- Declaraciones de familiares, compañeros de trabajo o amigos que hayan presenciado los episodios.
- Historial de medicamentos y evidencia de cumplimiento del tratamiento.
En Estados Unidos, las estadísticas de la epilepsia muestran su significativa prevalencia y el impacto que justifica la solicitud de beneficios por discapacidad:
| Estadística | Valor |
|---|---|
| Personas que viven con epilepsia | 3,4 millones (CDC) |
| Adultos con epilepsia activa | 3 millones |
| Niños con epilepsia activa | 470,000 |
| % incapaz de controlar las crisis con tratamiento | ~30% |
| Prestación mensual media del SSDI (2024) | $1,537 (media para todas las discapacidades, EE.UU.) |
La Importancia del Asesoramiento Legal Especializado
Solicitar la discapacidad puede ser un proceso complejo. Las solicitudes a menudo se deniegan por falta de evidencia, documentación incorrecta o incomprensión de los criterios. Contar con el apoyo de abogados especialistas en discapacidad permanente puede marcar una diferencia crucial al recopilar pruebas médicas sólidas, trabajar con proveedores de atención médica, representar al solicitante en audiencias o apelaciones y asegurar que la solicitud se presente correctamente, aumentando las posibilidades de obtener el apoyo financiero y la tranquilidad merecidos.
Consideraciones Generales sobre la Epilepsia
La epilepsia es una enfermedad cerebral crónica caracterizada por crisis epilépticas no provocadas y recurrentes, que afectan a más de 50 millones de personas en el mundo. La Organización Panamericana de la Salud/Organización Mundial de la Salud (OPS-OMS) la ha declarado una patología frecuente, mayormente tratable, pero que impacta severamente la calidad de vida y es altamente estigmatizante. Puede presentarse en cualquier momento de la vida, sexo o nivel socioeconómico.
Las causas más frecuentes incluyen traumatismos craneoencefálicos, infecciones del sistema nervioso central, problemas perinatales, accidentes cerebrovasculares y causas genéticas. El diagnóstico es esencialmente clínico, realizado por un neurólogo, con exámenes complementarios para precisar y clasificar el tipo de epilepsia. El tipo de crisis epiléptica (que es la manifestación clínica) depende de la región cerebral afectada, la velocidad de la descarga eléctrica, la etiología y la edad del paciente.
El pronóstico depende de la causa y del inicio y adherencia al tratamiento. Se estima que hasta el 70% de las personas con epilepsia pueden llevar una vida normal con el tratamiento apropiado, mientras que el 30% restante tendrá una epilepsia refractaria, requiriendo tratamientos complementarios como cirugía o dieta cetogénica. El uso de fármacos anticrisis es personalizado. Una crisis epiléptica se define como la aparición transitoria de signos o síntomas debido a actividad neuronal excesiva o sincrónica en el cerebro.
Las crisis epilépticas pueden ser:
- Provocadas: Consecuencia de una alteración orgánica conocida y aguda (p. ej., descompensación por abstinencia, fiebre en lactantes, privación extensa de sueño, hipoglucemia).
- No provocadas o espontáneas: Caracterizan la epilepsia, con inicio súbito, duración breve, estereotipadas (siguen el mismo patrón) y recurrentes.
tags: #epilepsia #mioclonica #es #invalidez